

成果简介
为析氢反应(HER)寻找高活性和低成本的电催化剂对于开发可持续能源至关重要,但这仍然是一个重大挑战。基于密度泛函理论计算,北京化工大学黄世萍、南京理工大学张胜利、新加坡国立大学郭翔宇等人报道了一种新型的双原子催化剂(HDAC),该催化剂具有嵌入g-CN的金属和非金属杂化中心。
计算方法
所有的自旋极化DFT计算都是在基于投影增广波方法的维也纳从头算模拟包(VASP)中进行的,并且交换关联势由广义梯度近似(GGA)中的Perdew–Burke–Ernzerhof(PBE)泛函描述。为了描述范德华(vdW)相互作用,作者采用了Grimme的DFT-D3方法,并分别采用3×3×1和6×6×1的Monkhorst-Pack k点网格进行结构优化和电子性质计算。
此外,作者将截断能设置为520 eV,能量和力的收敛标准分别设置为10−5 eV和0.01 eVÅ−1。为了避免周期性层相互作用,团队将真空层的厚度设置为15Å。溶剂化效应由嵌入VASPsol的隐式溶剂化模型来描述,其中水的介电常数为78.4。
结果与讨论

如图1a所示,作者选择三个可能的位点作为NM原子的结合位点,其中包括空腔中的中心位点(Ho)、与两个相邻的N原子结合的位点(I-2N)和与三个相邻的N原子结合的位点(I-3N)。
如图1b所示,NM(B、C、Si、P、S)与g-CN的结合能分别为−5.78、−5.60、−5.17、−3.81和−1.90 eV,表明这些系统是热力学稳定的。为了进一步验证它们的动力学稳定性,作者在500 K下进行从头算分子动力学(AIMD)模拟。经过5 ps模拟后的能量波动和结构变形(见图1c,d)证实了这些材料在热力学上是稳定的。

经过结构优化,作者发现氢原子吸附在NM的顶部,并向空腔轻微移动(见图2a),H和NM/g-CN之间的距离在1.09-1.49Å的范围内。与H在原始g-CN上的强吸附(在Ho位点上的吸附能为−4.39 eV)相比,NM的引入可以产生新的吸附位点来降低H的吸附强度。NM(B,C,Si,P,S)/g-CN的ΔGH*值(见图2b)分别为−2.26、−1.88、0.11、0.25和1.05 eV,表明H在非金属位点上的吸附强度是可调的。通过分析H吸附的PDOS(见图2c–g)可以发现,费米能级周围的H-s轨道和B/C/Si/P/S-P轨道发生杂化。

如图3a所示,作者考虑了除有毒和放射性元素外的所有3d、4d和5d金属原子(共26种金属原子),共产生了130种M-NM/g-CN HDAC。如图3b所示,128个系统(Zn-S/g-CN和Ag-S/g-CN除外)的形成能为负值,表明这些结构在热力学上是稳定的。除M-P(M=Zn,Y)/g-CN和M-S(M=Mn,Zn,Ag)/g-CN外,它们中的125个具有正的溶解势,这表明它们具有优异的电化学稳定性。基于稳定性标准,作者筛选出125种HDAC,以进一步探索其电子性质和催化活性。

如图4所示,在125个HDACs中,25种催化剂的ΔGH*值位于-0.19至0.19 eV,其中包括M-B/g-CN (M = Zn, Pd, Au), M-C/g-CN (M = Ti, Pd, Ag, Ir), M-Si/g-CN (M = Ti, Cr, Mn, Co, Ni, Zn, Rh, Ag, Ir, Au), M-P/g-CN (M = Fe, Ir, Au)和M-S/g-CN (M = Ti, Fe, Ni, Nb, Re)。
此外,11种催化剂的ΔGH*值与Pt(111)催化剂相当,即Pd-B/g-CN, Ti-C/g-CN, Ir-C/g-CN, Cr-Si/g-CN, Mn-Si/g-CN, Co-Si/g-CN, Rh-Si/g-CN, Au-Si/g-CN, Ir-P/g-CN, Fe-S/g-CN, and Ni-S/g-CN的ΔGH*值分别为−0.02, −0.03, −0.07, 0.05, −0.03, 0.07, 0.08, 0.01, 0.04, 0.08和−0.08 eV。
此外,ΔGH*为0.01eV的Au-Si/g-CN位于火山图的峰值附近,表现出优异的催化活性。位于阴影区域的10种HDAC也具有良好的催化活性,其中包括Pd-B/g-CN、Ti-C/g-CN、Ir-C/g-CN、Cr-Si/g-CN、Mn-Si/g-CN、Co-Si/g-CN、Rh-Si/g--CN、Ir-P/g-CN、Fe-S/g-CN和Ni-S/g-CN。

如图5所示,在PDOS中,H-S轨道和M-d轨道(NM-P轨道)之间存在明显的轨道重叠。H的价电子与M(NM)的活性价轨道相互作用,形成成键轨道和反键轨道。
为了定量描述键合效应,作者进行了COHP分析,Pd-B/g-CN、Ti-C/g-CN、Ir-C/g-CN、Cr-Si/g-CN、Mn-Si/g-CN、Co-Si/g-CN、Rh-Si/g-CN、Ir-P/g-CN和Fe-S/g-CN中M-H的ICOHP分别为-2.67,-0.34,-1.57,-1.70,-1.77,-1.61,-1.47,-1.35和-1.15 eV。Pd-B/g-CN、Cr-Si/g-CN、Mn-Si/g-CN、Co-Si/g-CN、Rh-Si/g-CN和Ni-S/g-CN中NM-H的ICOHP分别为-0.17、-0.68、-0.71、-0.67、-0.93和-3.00 eV。
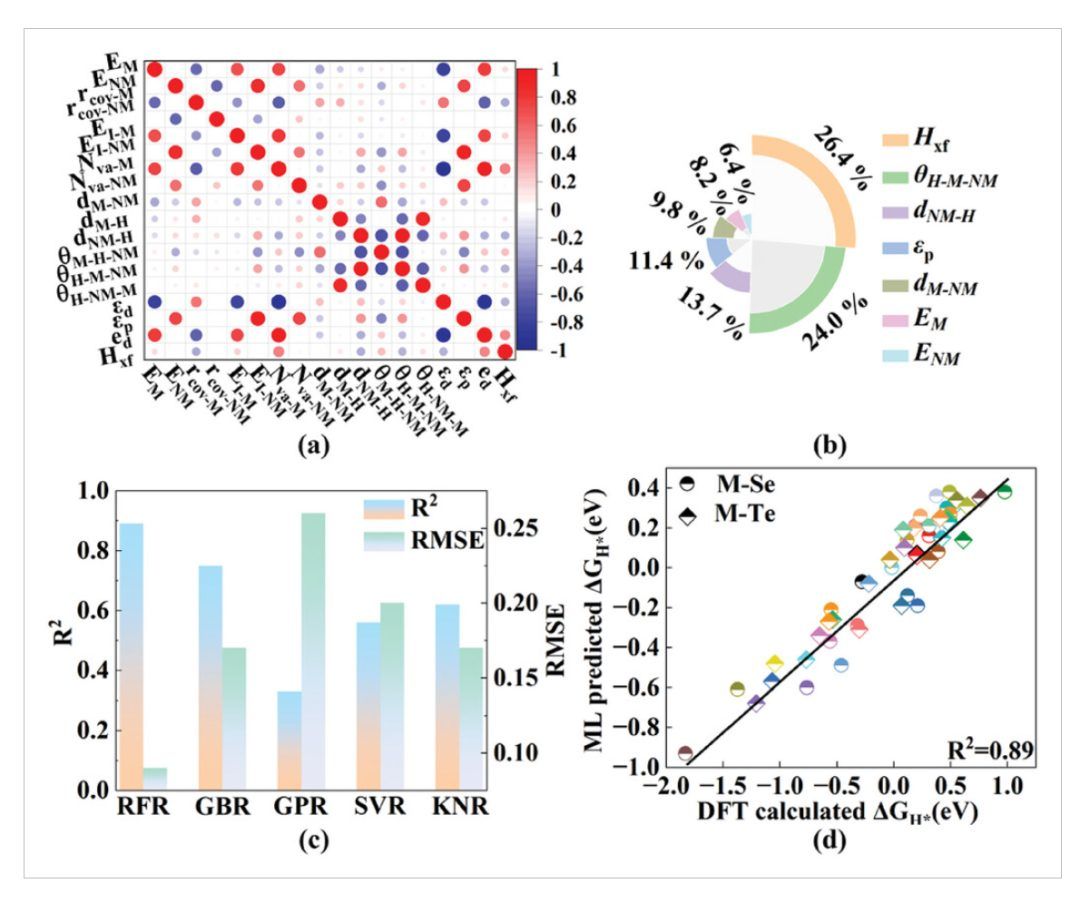
如图6a所示,EM、ENM、dM-NM、dNM-H、θH-M-NM、εp和Hxf被用作筛选ML模型的最终特征。如图6b所示,特征的重要性按降序排列为:Hxf(0.264)>θH-M-NM(0.240)>dNM-H(0.137)>εp(0.114)>dM-NM(0.098)>EM(0.082)>ENM(0.064)。
这一结果表明,金属原子的氢亲和力和氢的空间构型起着决定性作用,并且M-NM的配位环境协同影响催化活性。Hxf可以反映由于与Nva-M的强相关性而产生的电子增益/损失能力(见图6a),从而影响氢结合强度。H的两个空间坐标是由M和NM之间的相互作用决定的,其中包括θH-M-NM和dNM-H,,并且强相互作用会削弱H的吸附。
如图6c所示,作者比较了随机森林回归(RFR)、梯度增强回归(GBR)、高斯过程回归(GPR)、支持向量回归(SVR)、和k近邻回归(KNR)的稳定性和准确性。其中RFR具有最高的R2(0.89)和最小的RMSE(0.09),表明RFR模型适用于HER活性预测。
RFR预测的ΔGH*值如图6d所示,M(Sc、Ti、V、Cr、Fe、Zn、Ag、Ta、Re、Ir)-Se/g-CN和M(Sc、Ti、Mn、Fe、Co、Y、Nb、Mo、Pd、Ta和Ir)-Te/g-CN的ΔGH*值位于-0.20至0.20 eV。然后,作者对这52个HDAC进行DFT计算,并且证实M(Fe,Re,Ir)-Se/g-CN和M(Sc,Ti,Mn,Fe,Nb,Mo,Ir,Nb)-Te/g-CN具有良好的HER催化性能。
结论与展望
研究表明,在金属位点附近引入非金属原子(B、C、Si、P和S)可以实现它们之间独特的电荷转移,这为双原子中心提供了与单原子对应物截然不同的催化活性。在130种HDAC中,Pd-B、Ti-C、Ir-C、Cr-Si、Mn-Si、Co-Si、Rh-Si、Au-Si、Ir-P、Fe-S和Ni-S对是对质子具有近乎理想吸附强度的高性能电催化剂。机器学习分析可以直接识别影响催化活性的关键特征,并为快速筛选HDAC的未知化学空间建立可预测的框架。该工作为设计和开发潜在的HER催化剂开辟了一条新途径。
文献信息
Lihong Zhang et.al, Hybrid Double Atom Catalysts for Hydrogen Evolution Reaction: A Sweet Marriage of Metal and Nonmetal Adv. Energy Mater. 2023
