概述
药物分子设计前沿进展
分子编辑为药物合成及发现打开了新思路,通过在分子原有的结构基础上对其进行的原子尺度上的修饰,能极大程度地简化药物合成以及新药研发的难度。自其出现以来,化学家们主要围绕在两个方面不断优化:骨架编辑和外围编辑。

在这里给大家分享一篇最新的JMC期刊,对分子编辑技术进行总结并举例,为药物发现与结构设计提供新策略,如何开发一种普适的分子编辑策略也许会成为未来的主要研究方向。内容概览:
1. 骨架编辑:
1.1 单原子编辑
1.2 多原子编辑
1.3 扩换化
2. 外围编辑:
2.1 C-H键活化
2.2 原子/FGs交换
2.3 功能组转换
2.4 骨架重建
2.5 立体化学编辑
1 骨架编辑
近年来,骨架编辑因其充分发挥化学结构潜力而引发许多关注。骨架编辑以重排和骨架跃迁为中心,在光照、特殊试剂的作用下,通过单个或多个原子的删除和插入来进行分子骨架的调整。
1.1 单原子编辑
单原子编辑通过单原子操作直接转化核心结构,不会改变取代方式。作为药物设计中广泛使用的经典生物等构置换物,用N原子取代芳香族C-H基团可以有效地调整氢键相互作用、极性、溶解度、结合亲和力或对靶点的选择性。
用氮原子替换吡啶环中的一个碳,生成了FDA批准的药物阿伐那非,效价提高了20倍;TNNI3K抑制剂的开发也强调了该策略的重要性(图1a)。
2023年,Mark D. Levin的研究小组提出了一种碳氮转换反应,将喹啉类药物转化为相应的喹唑啉类药物,从而合成出了已批准药物talnetant的喹唑啉衍生物(图1a)。

Bill Morandi的研究小组实现了对硅烷基保护的吲哚进行骨架编辑,通过在五元环插入氮原子来引入喹唑啉或喹喔啉生物等排体,将β-肾上腺素受体拮抗剂吲哚洛尔转化为相应的喹唑啉衍生物(图1b)。

Sarpong实验室开发了α-酰化哌啶和其他饱和杂环的可见光诱导环收缩,通过该收缩,肽中的α-氨基酮直接转化为β-氨基酮(图1c)。

1.2 多原子编辑
多原子编辑能够通过特异性的方式将两个或多个原子插入分子骨架中,从而发现未知的药物化学实体。双环[2.1.1]己烷(BCHs)是一类重要的富含C(sp3)的苯类双环烃生物等排体,在药物化学中具有广泛的应用。
2023年,Procter研究小组报告了Sm(II)催化的分子编辑,通过将烯烃插入双环[1.1.0]丁烷片段,构建了BCHs这一重要结构(图2a)。利用该方法制备了一种新型广谱抗菌药物酞磺胺噻唑的衍生物。

Glorius及其同事发现了一种可见光介导的去甲基化环扩大策略,通过将双环[1.1.0]丁烷插入噻吩中的八元双环(图2b),该结构引入使得分子增强了与靶点的立体结合性。

1.3 扩环化
扩环化是一种新兴的分子编辑工具,一般指合成具有至少12个重原子的半刚性环,可以提供新的化学空间,与无环类似物相比可以提高对靶点的结合力或选择性,广泛用于编辑后续药物发现的药物(或临床候选)分子而无专利侵权。
Grubbs开发的闭环复分解(RCM)已成扩环化最常用的合成工具。RCM具有广泛的官能团适用性,并可适用于不同的反应需求。William及其同事利用RCM化学优化了大量专利的基于嘧啶的支架,生成了扩环口服激酶抑制剂Pacritinib(图3)

2 外围编辑
外围编辑主要关注在催化剂或其他条件下对惰性的C-H进行的活化,从而导入目的基团,实现在温和的条件下将目标分子官能化。
如:4′-修饰核苷用于抗病毒治疗的发现凸显了外周编辑的价值(图4)。4′-叠氮基-2-氟核苷阿兹夫定(也称为FNC)被NMPA批准用于HIV和COVID-19治疗。4′-乙炔基-2-氟核苷(CL-197)则是另一种高效、口服生物可利用的基于核苷的临床候选药物,用于治疗HIV-1感染。

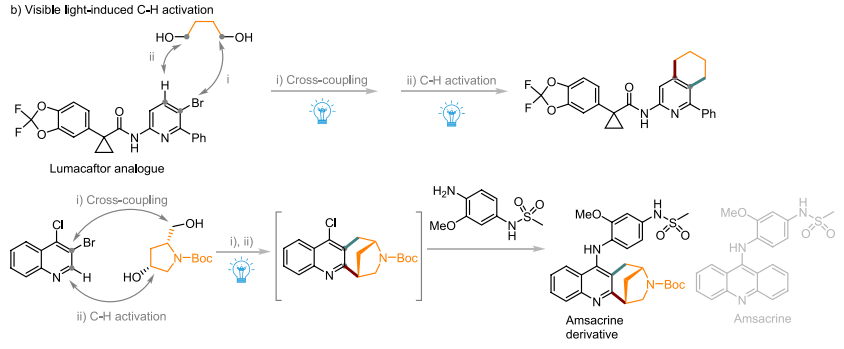
2.1 C-H活化
在结构优化中,通常会在分子支架上引入一个官能团,这可能会改变一些特性,如对靶点的活性和选择性。C−H键活化是一种强大的外周分子编辑策略,用于在不同位点高效选择性地功能化药效团。
Yu的研究小组开发了一种通用的氮杂芳烃的后期C−H烯烃化、烯丙基化和炔基化,实现了抗肿瘤药物喜树碱的C6或C7选择性烯烃化(图5a)。

MacMillan实验室开发了直接功能化的转化工具,通过融合镍催化的光氧化还原C−C交叉偶联和分子内C−H活化/功能化来构建各种非常难以进入的半饱和环系统,成功合成了抗囊性纤维化药物Lumacaftor和抗肿瘤药物Amsacrine(图5b)。
2.2 原子/FGs交换
将易反应的原子或FGs(如卤素、硼酸酯和羧基)转化为其他结构是一种常见的药物修饰策略。其中,过渡金属催化的交叉偶联反应常用于将卤素或硼酸酯基团转化为杂环或苯环,提供与靶点关键疏水或氢键的相互作用。
2017年,第一个氘代药物Deutetrabenazine,被批准用于治疗亨廷顿病相关的舞蹈症。Deutetrabenazine表现出比非氘化药物丁苯那嗪更好的药代动力学特性和有效性(图6a)。
2022年,TYK2抑制剂Deucravacitinib获批用于治疗中度至重度银屑病,发现氘化可避免体内N-脱烷基化代谢(图6a)。

其中还报道了一种光催化极性转化策略,即通过一锅法使用D2O作为氘源将异氰酸酯基团转化为氘代α-氨基氧化膦基团,产物表现出比相应的非氘化合物提高近10倍的效力(图6b)。

2020年,Lundgren及其同事报道了一种脱羧-羧化平台,通过非催化原子交换可合成13C同位素标记的非甾体抗炎药非诺洛芬和伊索克酸(图6c)。

2.3 功能组转换
鉴于FGs的连接位置对药物分子的各个方面,包括生物活性和物理性质有显著影响,FGs转化的发展在药物发现中具有特别的意义。
2021年,Dong课题组报道了1,2-羰基迁移法,分两步将醋酸二氢睾酮转化为“C2-oxo”类似物,收率为51%,具有良好的区域选择性(图7)。原合成需5个步骤,收率较低(< 39%)且缺乏区域选择性。

2.4 骨架重建
骨架重建是药物化学中一种有吸引力的策略,涉及将一个简单的杂环转化为具有相同环大小但FGs更多的骨架结构。
在降胆固醇药物阿托伐他汀(立普妥)的研究中,将二取代吡咯转化为完全取代吡咯提高了抑制活性 > 30倍(图8a)。

2023年,Zhou及其同事报告了一种一锅方法,通过去甲基化解构和重构过程,以偶氮烯烃为分子微扰剂,将简单的吡咯转化为完全取代的吡咯(图8b)。

2.5 立体化学编辑
手性生物活性化合物的立体化学通常影响其效力、药代动力学、药代动力学等。立体化学编辑可以在后期实现立体中心的相互转化,并降低化学合成的难度。
Wendlandt研究小组的开创性工作揭开了一种新型的光诱导立体编辑逻辑,将具有完全未活化的三级立体中心的多样手性分子转化为截然不同的立体异构体。

总结
分子编辑策略颠覆性地改变了传统药物发现和开发方式:
通过常规方法获得未知的分子结构,发掘药效官能团作用;
以直观和精确的方式快速构建药效结构;
选择性地对分子骨架的位点引入不同的官能团,从而加快化学制备;
实现分子结构的多样化,丰富化学库。
当然除了以上诸多优势外,也要注意:对复杂分子骨架进行分子编辑,有时会导致添加不必要的取代基而对药效或选择性方面产生不利影响;另一个问题是针对性地编辑某个位点很大程度上取决于使用的反应条件和试剂,还需要化学家的进一步探索。
END
文案 | 李怡娴
排版 | 李怡娴
审核 | 姜笑南
发布|姜笑南
世界生命科学大会
RECRUIT
关注我们,获取生命科学
学界前沿|促进更多的学术交流与合作
业界前沿|促进更快的产品创新与应用
政策前沿|促进更好的治理实践与发展
我们期待你的加入
