如果说10月8日颁发的诺贝尔物理学奖是与人工智能有点暧昧不清,那么10月9日揭晓的诺贝尔化学奖就彻底“不装了”,看来ChatGPT拿文学奖也指日可待了(bushi)。
2024年的诺贝尔化学奖授予大卫·贝克尔(DavidBaker),以表彰其计算蛋白质设计方面的研究;以及德米斯·哈萨比斯(DemisHassabis)和约翰·詹珀(JohnJumper),以表彰其在蛋白质结构预测方面的研究。

这两项研究都与人工智能与生物学的深度结合有关。蛋白质设计和蛋白质结构预测方面的工作,若非人工智能的加持,注定是一个极其有意义但同时极其无解的领域。
下面,我们就来一起了解一下,为什么这项工作意义重大而又如此困难,人工智能又为解决这一问题作出了怎样的贡献。
蛋白质的功能与结构
蛋白质是一切生命活动的体现者。大脑传递信息,靠的是蛋白质搬运电荷;肌肉收缩,靠的是蛋白质互相拔河;细胞需要能量,靠的是蛋白质运输氧气。癌症、阿兹海默症、艾滋病、糖尿病……几乎任何疾病都与蛋白质有着千丝万缕的联系。
换句话说,想要研究透彻生命,就必须研究透彻蛋白质;想要研究透彻蛋白质,就必须完全了解其结构的秘密。
很久之前人们就知道,蛋白质虽然种类可能上亿,形态结构千变万化,但是它们大部分都是由20种组成元件拼成的,这些元件就是氨基酸。就像乐高积木虽然能拼出形形色色的物体,但是其基础小块的数量却很少。
一个蛋白质到底选用了哪些氨基酸以及它们按顺序排列成多肽链,这被称为蛋白质的一级结构;这些氨基酸排好序之后,它们之间能形成哪些基本构型,这被称为蛋白质的二级结构;这些构型之间如何相互靠拢、形成有功能的团块,这被称为蛋白质的三级结构;有的蛋白质还需要几条多肽链之间的相互结合,这被称为蛋白质的四级结构。
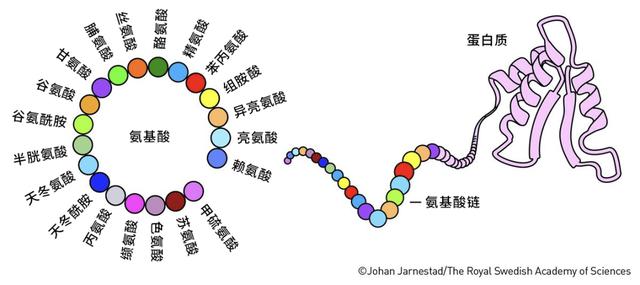
人们发现,蛋白质的复杂功能,完全依赖于蛋白质的三级和四级结构。蛋白质从线性的多肽链形成复杂却精巧的三维结构,这就是蛋白质折叠。生物中的每个蛋白质都是极致的平衡大师。它们的形状、大小、亲水性、电荷数量等特性被严密地控制在一个最适合的范围内,从而保证它们各自能够行使不同的生物学功能。
而这些高级结构能够维持,又是由于一级结构所决定的。一级结构如果出错,那么就会导致所有的高级结构统统错误,严重情况下会导致整个蛋白质失效。
例如,如果在血红蛋白的某个位置上,本来亲水性的氨基酸突变成了不亲水的氨基酸;这个不亲水的氨基酸会导致这个血红蛋白的亲水性大大降低,于是它们不是溶解在血液里,而是互相凝固在一起。凝固在一起的血红蛋白无法携带氧气,这就是镰刀形红细胞贫血病的病因。
因此,蛋白质的一级结构是基础,而它的高级结构是表象。那么,我们是否可以从基础推测表象,或者依据想要实现的表象,而设计出基础呢?
要了解蛋白质结构的秘密,太难了
一级结构的测定方法早在上世纪七十年代就已经问世。而从上世纪五十年代开始,化学家们就已经能够根据氨基酸的性质,推测相邻氨基酸之间的相互作用,以及它们能够形成的构型,因此人们对二级结构的了解也很深入。
此时,人们已经确信,蛋白质的高级结构,完全由其氨基酸序列确定。但这也带来了一个悖论:氨基酸序列能够产生的排列可能是一个天文数字,如果细胞把每个可能性都尝试一遍,那可能要到宇宙毁灭那一天。换句话说,蛋白质是如何快速“选择”自己该折叠成什么结构的呢?其背后的规则是怎样的呢?
一旦掌握了这个规则,那么我们就能解决两个重要问题:一是蛋白质结构预测,二是蛋白质结构设计。
换句话说,一旦掌握了蛋白质折叠的规则,那么人类就成为了主宰蛋白质世界的“神”,我们就可以快速解读出世界上任何蛋白质的功能,从而清晰地认识生物;也可以任意向生物中添加某种功能的蛋白质,从而定向地改造生物。
这么重要的规则,其难度当然可想而知。
用乐高的比喻来说,这两个问题就是:如果给我一堆乐高零件,我能不能预测一下它们能拼成什么物体;如果让我去拼一个物体,我能不能徒手画出它的设计图,并且判断哪种设计图最省时省力。
对于乐高来说,各个小块之间的相互作用是确定而稳固的,它们互相可以卡住,并且只能从固定的方向卡住。
但是对于氨基酸来说,它们可能以许多类型的力进行相互作用,例如电荷相互作用、疏水作用、氢键等。更要命的是,这些氨基酸在空间上可能以任意的角度、方向和距离进行相互作用;而且相互作用的大小、类型都会为不同的功能来服务,并不是一成不变的。
起初,人们希望通过解构一些有代表性的蛋白质的结构,对蛋白质折叠规则来个“管中窥豹”。结果窥了好几百次豹才发现,每次窥到的东西都不一样,有时候还不能确定窥到的是不是豹子身上的斑点。
毕竟,蛋白质的功能有几十万种,结构有几百万种,它们内部到底隐藏着多少规则,这已经远远超出了人脑的理解能力范围。
利用人工智能的强项解决问题
然而,这种从大量重复中寻找固定模式,并且引用这一模式来解决实际问题的工作,恰好是人工智能的强项。
人工智能能够将蛋白质的几十万种功能和几百万种结构一个个地学习并总结起来,它虽然不能向人类描述其中的折叠规则,但是却能够合理地运用它所发现的规则,从而间接实现蛋白质结构设计和结构预测。
下面就是今年获奖者们的一些尝试。
1999年,贝克尔和同事开发了Rosetta计算机程序。这一程序先是学习了当时人类已知的蛋白质数据库,然后使用蒙特卡洛优化,主要考虑范德华相互作用、氢键和溶剂化效应等因素,从而给出几个可能得方案。
贝克尔用这一程序设计了一个特定功能的蛋白质,程序给出的方案与任何自然界中存在的蛋白质都不同;人们将它合成出来之后,确实发现它能够行使贝克尔预先设想的生物学功能。这也是人类首次成功设计出复杂功能的蛋白质。
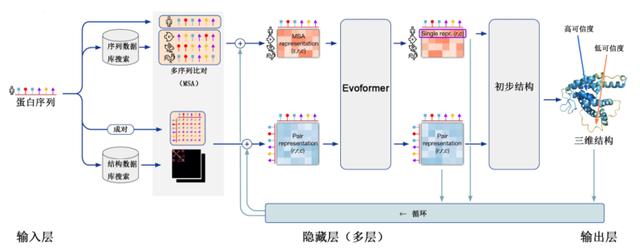
时间到了2018年,哈萨比斯和詹珀的Deepmind公司,开发了基于卷积神经网络的人工智能AlphaGo,通过学习了上万亿盘围棋之后掌握的围棋规则,彻底击败了人类;于此同时,Deepmind公司将这种快速学习的人工智能应用于蛋白质结构预测领域,开发了AlphaFold,预测成功率达到了60%。
到了2020年,AlphaFold的升级版AlphaFold2的预测结果已经可以做到与实验测量结果几乎没有误差,也就是说只要告诉它蛋白质是由哪些氨基酸构成的,那么它就会告诉你这个蛋白质的结构和功能是怎样的。而2024年发布的AlphaFold3虽然精确度变化不大,但具有了一定的通用性,不再局限于蛋白质,还可以用于其他生物高分子与小分子配体、高分子修饰,以及蛋白质和它们的复合物结构。
结语
可以说,贝克尔、哈萨比斯和詹珀的工作开辟了生化和生物学研究的新时代,我们现在可以用以前无法想象的方式预测和设计蛋白质结构。
当然,如果没有结构生物学家的努力,上述进展不可能实现。他们为蛋白质数据库提供了大量实验确定的蛋白质结构。这些数据是数十年蛋白质结构测定研究的结果,为今年获奖者在蛋白质设计和结构预测方面取得的决定性突破奠定了基础。
有人也许会问“人工智能这么强大了,结构生物学家是否会失业?”其实,就像ChatGPT没有让写手失业一样,人工智能在蛋白质设计和结构预测方面还有很多问题有待解决(比如对复合体、柔性区的预测等等)。对于结构生物学家来说,“获得结构”只是研究的手段,理解生命,做出生物学发现才是目的。正如颜宁院士所说,“如何能够理解我们细胞里各个分子的动态变化,是我们目前面临的最大挑战之一”。
