导语
茶是世界上消费最广泛的非酒精饮料,因为茶叶富含儿茶素、茶氨酸和咖啡因等特征化合物,对健康有诸多益处。尽管测序技术不断进步,但茶叶关键农艺性状的遗传机制仍不清楚。基因组辅助育种技术有望加速主要作物的育种,但育种工作的可持续性有赖于不断发现与关键农艺性状相关的关键基因的有益等位基因。与只保留一个单倍型的单一参考基因组相比,泛基因组能以单碱基分辨率捕捉物种内部遗传变异的全貌。近日,有学者在《Nature Plant》上发表了题为“Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia Sinensis”的最新茶树泛基因组研究成果。让我们一起来学习一下吧~
文章背景

文献标题:Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia Sinensis
发表期刊:Nature plants
影响因子:18.0
发表时间:2023.11.27
研究思路
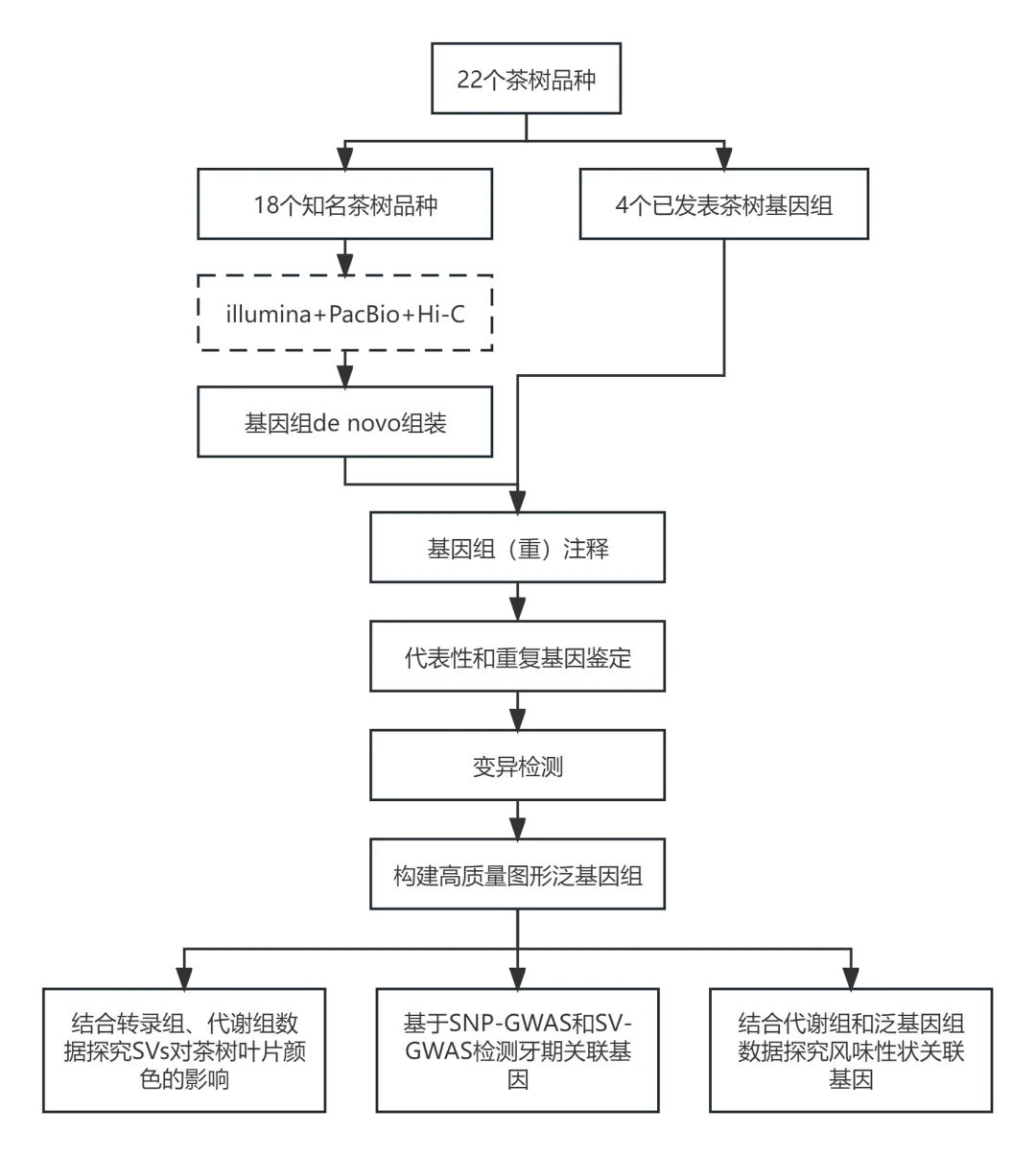
研究结果
1、多个优良茶树品种的重新组装和注释
选择了18个知名的茶树品种进行基因组测序和从头组装,这些品种分布在所有三个中华茶树类群中(图1a,b)。加上先前发表的4个茶树基因组(LJ43、SCZ、HD和TGY),该研究共分析了22个基因组,充分代表了中华茶树的广泛遗传多样性。基因组组装的结果显示,这些茶树基因组大小相似,除了少数倒位和易位外,大多数基因组区域具有高度一致性。注释结果显示茶树基因组中有高达5万至6万个蛋白编码基因,相较于已发表的版本,编码基因数目增加了近20%。茶树基因组包含大量的重复序列,其中长末端重复转座子(LTRs)占据了基因组序列的50%以上。茶树基因组一次近期的LTR爆发事件(LTR burst,约30-50万年前)导致蛋白编码基因的迅速扩张。
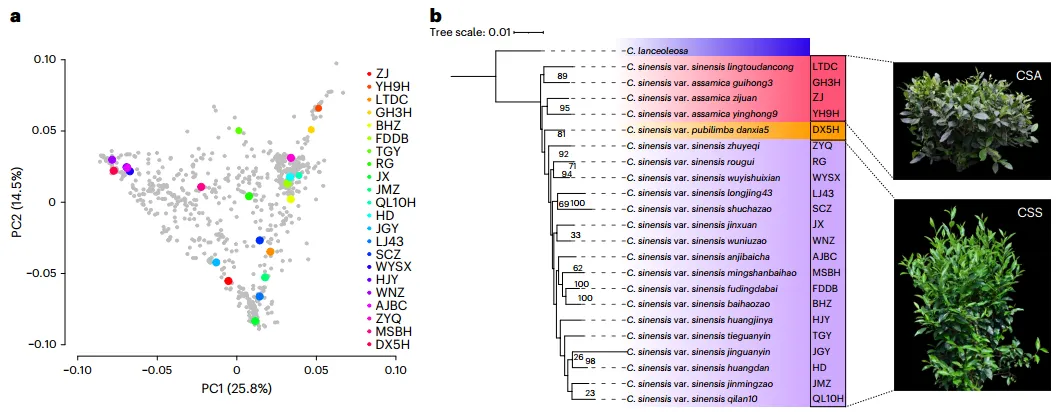
图1 22个泛基因组测序的代表性茶树品种
2、由TE驱动的茶基因组结构变异(SV)
通过22个基因组之间的两两比较分析,任何两个样本之间的基因变异平均约为7.6%。将剩余的21个基因组与TGY参考基因组进行比较,共发现了887,986个SVs均匀分布在15条染色体上(图2a),包括435,505个缺失、421,642个插入、6,595个重复和24,244个倒位(图2b)。5 Mb滑动窗口分析显示,79.8%的SV与各自区域中的转座子TE重合,SV与TE之间存在明显的相关性(图2c)。这些结果表明TEs在驱动遗传变异方面起着关键作用。为了协调不同基因组中的等位基因,生成了一个包含40,461个代表性基因和432,597个等位基因的泛基因表,包含29.8%的core基因、43.9%的shell基因和26.3%的private基因(图2d)。Core等位基因的编码序列(CDS)较长,Ka/Ks明显较低,表达量也高于dispensable等位基因,这表明核心基因在功能上是保守的(图2e-g)。
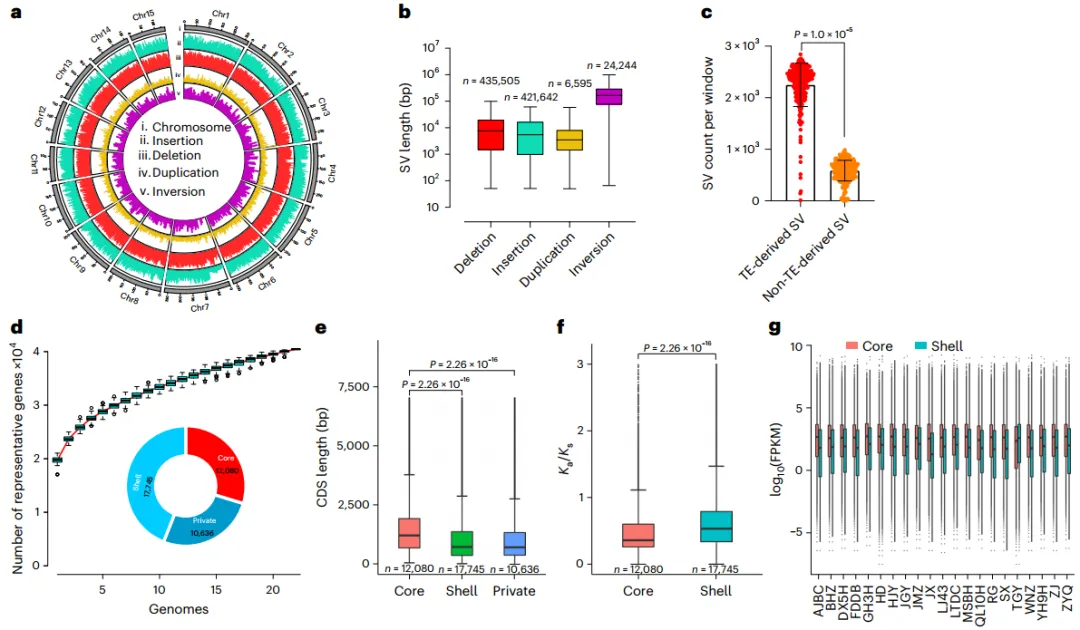
图2 基于22个从头组装茶基因组的SVs检测及泛基因组构建
3、结构变异对茶树不同叶片颜色的影响
收集具有重要经济价值的不同栽培品种的茶树转录组和代谢数据。研究显示,不同品种之间的叶片颜色差异可能归因于与这些途径相关的基因的SVs。通过对相关的基因进行序列比对,在7个关键酶中发现了多个SV(图3a,b)。值得注意的是,‘AJBC’和‘HJY’品种的GluRS基因显示了反密码子识别结构的缺失,可能导致谷氨酰-tRNA合成酶功能受损,从而影响叶绿素合成。此外,CYP97A3HJY等位基因在不同叶片颜色的样品中表现出不同的突变(图3b)。RNA-seq分析还显示CYP97A3HJY等位基因在“HJY”中高表达(图3c,d),说明其对玉米黄质积累的潜在贡献。值得注意的是,6个编码假定转录因子的基因的转录本丰度在紫色叶片的样品中明显较高(图3e)。虽然这些基因的序列比对显示它们的CDS整体差异不大,但确实在CsMYB114的上游约 200 bp 处发现了一个148 bp的LTR/Gypsy插入,该插入只存在于紫色叶片的品种中(图3f,g)。因此,该研究结果表明,这些SVs可能参与了茶叶颜色的调控。
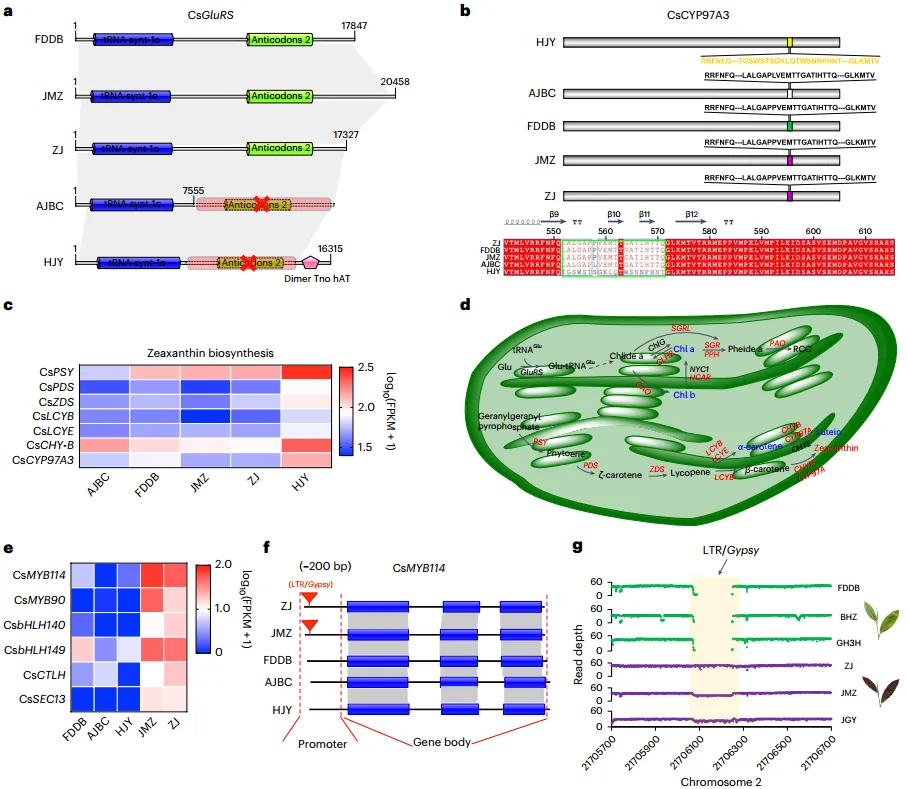
图3 与茶叶颜色调节相关的SVs功能影响
4、基于Pan-GWAS的茶树芽期相关基因挖掘
基于155个重测序样本进行的GWAS发现了35个与早花芽分化(EBF)性状显著相关的SNPs(图4a)。Pan-GWAS分析在特定基因座中发现了一个SV与DREB2A-like基因的上游区域重叠(图4a,c),该基因在EBF和晚花芽分化(LBF)品种之间也有显著的表达差异。将不同品种的DREB2A -like等位基因进行比对,发现了一个A到C的非同义突变,该突变将一个氨基酸从EBF品种中的异亮氨酸改变为LBF品种中的亮氨酸(图4b)。这一突变改变了该DREB2A-like蛋白的推测三维蛋白结构,并可能影响基因功能。DREB2A-like CDS系统进化树将EBF与LBF区分开来(图4d)。该结果表明,DREB2A-like等位基因可能对控制茶叶的EBF性状有影响。

图4 茶树泛基因组GWAS挖掘
5、茶泛基因组使基因组学辅助育种成为可能
代谢组和茶泛基因组数据显示,共有495个至少含有一个突变的基因与儿茶素含量有显著相关性(图5a)。功能分析显示,这些基因有几个是以前报道过的参与类黄酮生物合成或儿茶素合成的基因。其中,在CsCYB-561、CsWDR和CsDELLA基因中检测到的单碱基替换与儿茶素含量显著相关(图5b)。此外,还发现位于CsDIOX基因氨基末端序列(A-A至G-G)的两个相邻非同义突变导致丝氨酸(S-S型)被甘氨酸(G-G型)取代。这些突变与泛基因组和整个茶叶群体中的儿茶素含量高度相关(图5c、d)。进一步分析表明,CsDIOX位于3号染色体(CGC3)上的一个基因簇中,该基因簇具有合成儿茶素的潜力。如图5e所示,该基因簇在山茶属植物中是保守的,由六个核心基因组成。在这六个基因中,CsSCPL4和CsSCPL5被认为是合成儿茶素所必需的基因。
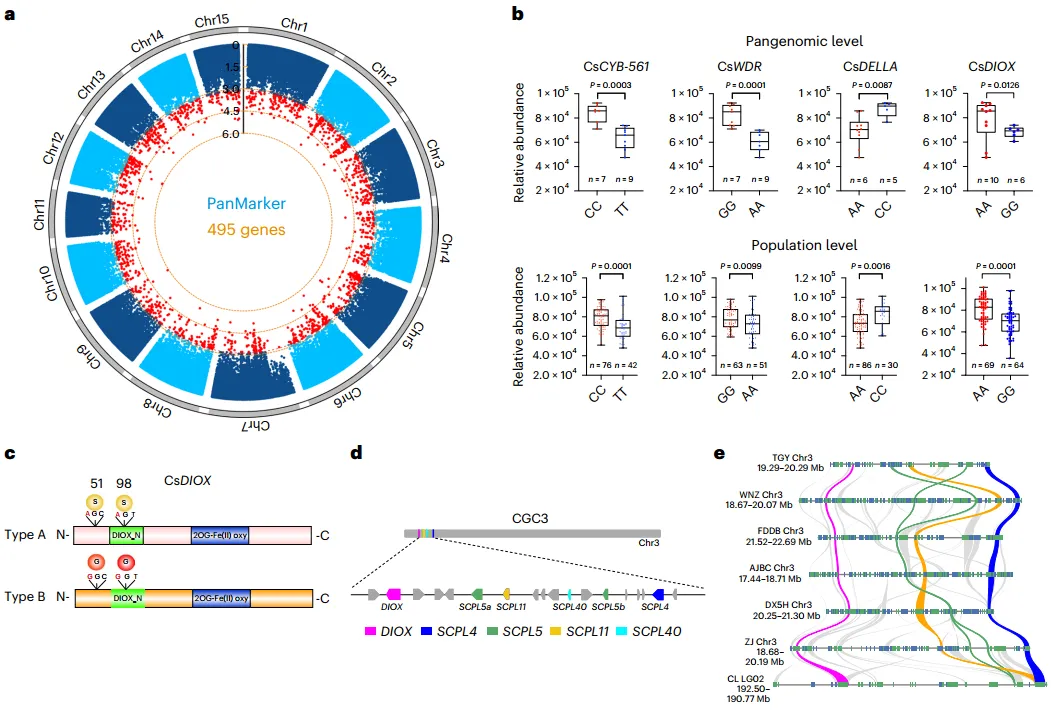
图5 泛基因组使基因组辅助育种成为可能
研究结论
该研究通过对22个典型茶树品种的基因组测序和泛基因组组装及构建,揭示了茶树基因组的庞大及复杂性,强调了泛基因组在揭示关键基因方面的优势。研究发现茶树基因组中蛋白编码基因数量之多超过预期,主要源于小规模的基因重复,并强调了结构变异在叶片颜色调控和早芽萌发等复杂性状中发挥的关键作用。总之,该研究推动了对茶树进化和遗传学的深入理解,为茶树产业的可持续发展和改良提供了科学依据,也为其他非模式植物的基因组研究提供了参考。
参考文献
Chen S, Wang P, Kong W, Chai K, Zhang S, Yu J, Wang Y, Jiang M, Lei W, Chen X, Wang W, Gao Y, Qu S, Wang F, Wang Y, Zhang Q, Gu M, Fang K, Ma C, Sun W, Ye N, Wu H, Zhang X. Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia sinensis. Nat Plants. 2023 Dec;9(12):1986-1999. doi: 10.1038/s41477-023-01565-z. Epub 2023 Nov 27. PMID: 38012346.
