

传统的 Kölbel-Engelhardt 合成方法通常在高温和高压条件下将一氧化碳和水加氢生成碳氢化合物。虽然已经证实了由光驱动CO加氢生产碳氢化合物可以在较温和的条件下进行,但这个反应必须首先获得H2。
基于此,北京大学马丁教授、郑俊荣教授、中国科学技术大学刘进勋教授以及上海交通大学刘晰副教授(通讯作者)等人共同报告了一种在常压下通过光驱动的方法,直接从一氧化碳和水合成碳氢化合物,无需外部加热或添加氢气。研究使用了TiO2载体上的镍催化剂,获得了较高的活性和C2+选择性。原位光谱和密度泛函理论计算结果表明,光生电子从二氧化钛向镍的迁移促进了碳-碳耦合反应,在二氧化钛-镍催化剂界面上观察到了多碳产物高选择性的原因。
研究背景
最近催化CO加氢生产液体燃料引起了广泛的研究,这个过程也被称为费托合成(FTS)。人们对于从废碳资源和生物质中生产有价值的碳氢化合物的兴趣再次被激发。每年工业部门产生数十亿吨的废CO,而这些废碳资源通常被燃烧成二氧化碳或直接释放到环境中,利用这些废CO资源生产高附加值化学品或液体燃料可能在成本上具有优势。
此外,利用废碳资源还可能是减少碳排放的有效途径,研究人员对于开发利用废碳资源的方法具有浓厚的兴趣。
图文导读
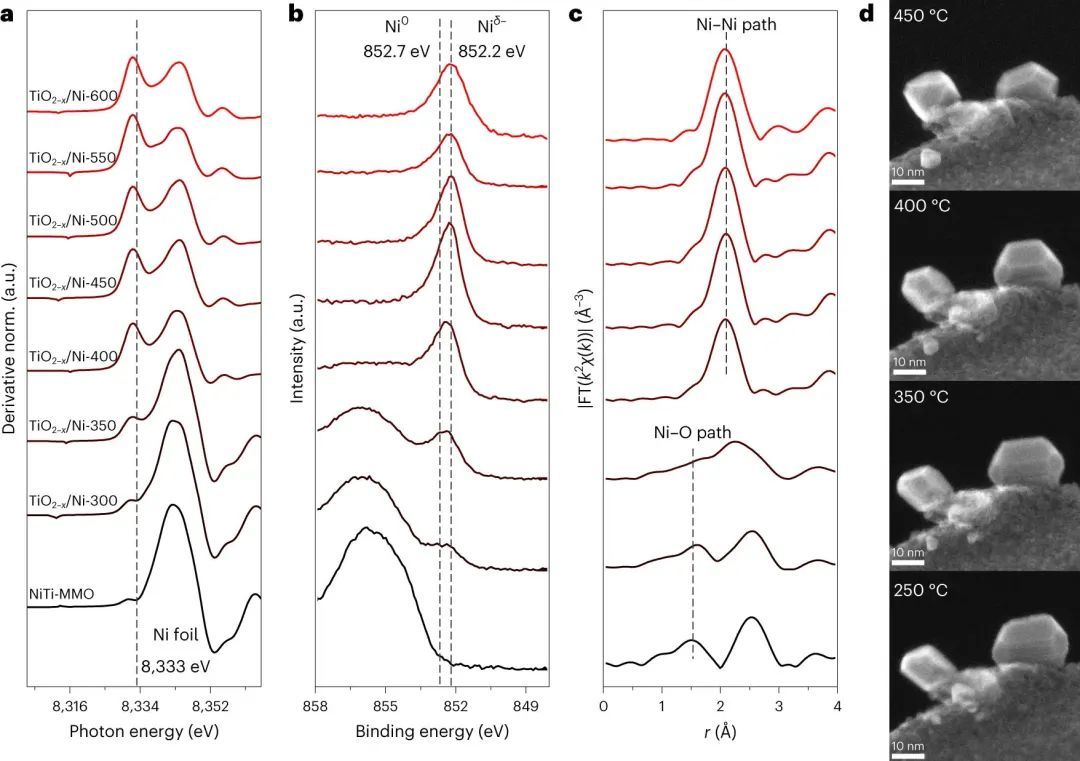
本文采用水热法合成了NiTi层状双氢氧化物(LDH)纳米片。将NiTi-LDHs在500℃下煅烧4 h,制备了由NiO和TiO2-x组成的混合氧化物(NiTi-MMO)。利用准原位X射线衍射(XRD)、X射线吸收光谱(XAS)、紫外-可见-近红外(UV-Vis-NIR)分光光度法和X射线光电子能谱(XPS)分析了在不同温度下还原的TiO2-x/Ni催化剂的结构(T为催化剂的还原温度,表示TiO2-x/Ni-T)。在400°C时,Ni被还原为金属态(图1a-c)。在450°C时,TiO2−x/Ni-450催化剂的Ni吸收边转移到比Ni箔更低的能量,这表明存在比金属态更低价电子的Niδ−种。
随着还原温度的升高,吸收边及其一阶导数逐渐向较低的能量迁移(图1a)。从准原位XPS结果(图1b)来看,在低于350℃的还原温度下,2p3/2光谱中氧化镍的峰占主导地位,而在350℃或更高的还原温度下,852.7和852.2 eV的峰成为主要峰,这分别是由于Ni0和Niδ−物质的存在。即使在较高的还原温度下,852.2 eV的峰也占据了光谱的大部分,表明进一步还原的催化剂中形成了更大比例的Niδ−物质。此前有报道称,Ni和TiO2−x之间的强金属-载体相互作用(SMSI)会使电子从TiO2−x载体转移到Ni,这解释了Niδ−物种的电子特征。
与Ni-TiMMO主要在紫外区表现出强吸收不同,TiO2−x/Ni-T在整个光谱中表现出广泛的吸收,表明TiO2−x/Ni-T具有全光谱吸收能力。不同光强下TiO2−x/Ni-450催化剂床层的表观温度在202℃~248℃之间。
有趣的是,TiO2−x/Ni-450催化剂的Ni 2p壳层原位XPS谱显示,依次在点亮和熄灭条件下,Ni 2p3/2峰在再照射条件下移动到一个更低的束缚能,随后在熄灭条件下又回到原来的位置,这说明了光照射诱导Ni表面的可逆电荷积累。这意味着镍在光催化过程中可以作为电荷受体/供体。环境干与二次电子探测器被用来证明镍基催化剂的固有的物理化学行为。将钝化后的TiO2−x/Ni-T催化剂在H2气氛(压力10 Pa)中不同的还原温度下还原1 h,从250℃到450℃,随着还原温度的升高,Ni纳米颗粒的形貌逐渐变为规则的截尾八面体;同时将Ni纳米粒子嵌入到TiO2−x中(或TiO2−x向Ni纳米粒子迁移)(图1d)。上述验证结果充分说明了Ni纳米粒子与TiO2−x之间的强相互作用。
表1. 镍基催化剂的光热催化性能

然后,作者评估了TiO2−x/Ni催化剂在常压光照射下催化CO和H2O转化的性能,结果总结在表1中,没有催化剂在光照射下没有产物(表1,条目1)。此外,氧化物前驱体(NiTi-MMO)的CO转化率很差(1.7)。相比之下,还原的催化剂表现出相当大的活性,并且活性对还原温度也有很强的依赖性。WGS在光热催化中的关键作用可以通过在所有还原的催化剂中产生可观的CO2和H2来证明,但后续CO加氢活性随着Ni的还原而出现并增加。TiO2−x/Ni-300的金属Ni含量较低,CO转化率为9.8%,主要产物为甲烷(表1,条目2)。
从300℃到450℃,随着还原温度的升高,CO转化率和C2+选择性均逐渐提高(表1,条目2 - 5),随后C2+选择性超过甲烷选择性。表明金属Ni甚至Niδ−是光驱动级联KES反应的活性位点。当还原温度从450°C升高到600°C时,CO转化率达到最大值,随后下降,这可能是由于高温还原后暴露的Ni位点数量减少,可能是由于Ni物种的电子结构进一步变化(表1,条目5-8)。
优化反应条件后,TiO2−x/Ni-450催化剂在辐照条件下的成烃活性为8.83 mol−CH2−molNi−1 h−1,C2+选择性为55.5%(表1,条目9),甚至超过了文献报道的热催化KES反应的性能。更有趣的是,TiO2−x/Ni-450催化剂在模拟阳光下也具有活性(表1,条目10)。2 h内CO的转化率为8.1%,C2+的选择性达51.9%以上。

到目前为止,由于反应体系复杂以及光子作用难以明确,大多数光热催化过程的机理尚不清楚。这个问题已经用原位光谱实验进行了研究。在光催化或光热催化反应中,学者们广泛关注催化剂材料被光子激活后的电子转移过程。然而,在光热过程中,对催化剂中电子迁移的直接观察仍未实现。
为了进一步了解光催化或光热催化反应过程的机理,迫切需要超快响应的原位表征。本研究采用原位时间分辨漫反射傅立叶变换光谱(DRIFTS)来研究CO的活化和中间体形成的反应路线(图2a,b)。更重要的是,作者使用原位超快红外光谱(图2c-f)来识别各种催化剂上的短寿命瞬态物种,并区分它们与光子激发相关的光催化行为,这可以帮助我们更好地理解光催化和光热催化之间的区别。
图2a和b中~ 2075/2071 cm−1和~ 1925/1923 cm−1的红外波段分别被分配给Ni缺陷位点上的线性吸附CO和Niδ−位点上的桥式吸附CO,它们都是在CO和H2O吸附后的光热或热条件下观察到的。在~ 2941 cm−1处(CHx*种的不对称vs(C-H)伸缩振动)和~ 2886 cm−1处(CHx*种的对称vs(C-H)伸缩振动)的红外波段以及CHx*种的弯曲振动对应的~ 1378 cm−1和~ 1354 cm−1处的红外波段随着辐照时间的增加而逐渐增加(图2a)。
此外,在3016 cm−1处(分配给CH4分子的v(C-H)拉伸振动)没有发现明显的IR波段。这说明光热催化的主要产物是C2+烃类化合物,而不是CH4。令人惊讶的是,作者发现与热催化相比,光热条件下存在特殊的反应中间体(1600 cm−1至1100 cm−1属于C-O和C-C振动),除了产物变化,这表明光催化过程可以改变传统的化学路线,允许产生更多的碳氢化合物。
此外,本文利用原位可见泵浦/IR探针光谱研究了这种光子诱导的电子激发和转移过程,该过程在光热反应中发挥了至关重要的作用。纯TiO2表现出典型的冷却和俘获过程(图2d),而Ni/Al2O3表现出微弱的信号和自由电子的快速衰减过程(图2f),表明在520 nm激发下,自由电子的激发来自TiO2而不是Ni。
与Ni/Al2O3和TiO2相比,TiO2−x/Ni-450催化剂表现出明显更长的弛豫过程(图2c-f),这表明电子从TiO2−x转移到Ni,导致TiO2−x的捕获过程更加困难。在TiO2−x/Ni-450催化剂的光子诱导电子激发和弛豫过程中(图2e),在520 nm激发处出现了~ 2170 cm−1和~ 2120 cm−1两个吸收峰,这两个吸收峰属于位于Ni上不同位置的CO的激发态吸收。
这两个峰与它们各自的基态吸收峰在傅里叶变换红外光谱中分别位于2174 cm−1和2120 cm−1的位置相比(图2a),由于电子激发引起了轻微的红移,这表明自由电子向CO转移,这也得到了作者的DFT计算的支持。Ni活性位点上CO振动弛豫的出现表明,在辐照下TiO2−x向Ni发生电子转移过程后,自由电子从金属Ni向CO转移。因此,作者得出结论,自由电子激发可能起源于TiO2,然后转移到Ni,最终转移到CO。
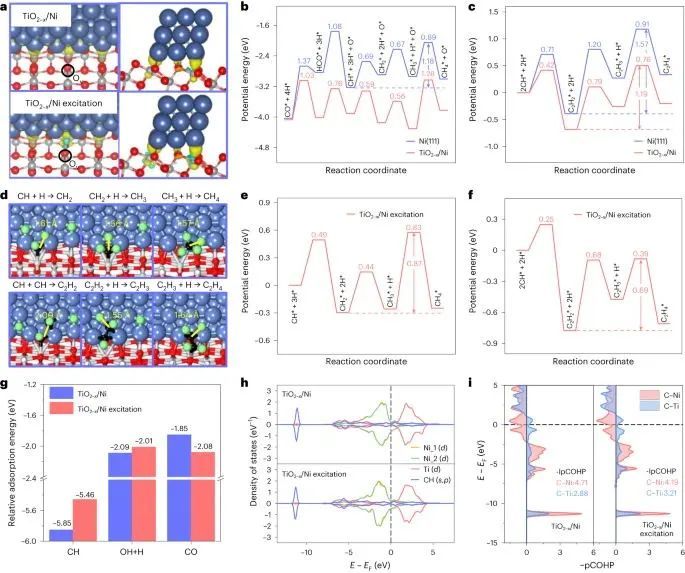
在TiO2−x/Ni催化剂上,Ni纳米晶体被支撑在部分还原的TiO2衬底上,通过DFT计算来理解CO加氢机理。界面TiO2−x/Ni催化剂通过使用由三列Ni原子吸附在缺氧TiO2上的三层Ni带来模拟(101)(图3a)。Ni(111)表面首先作为参考进行研究,因为它均匀地暴露在金属Ni纳米颗粒上(图1d)。在光驱动的热催化过程中,通过HCO形成CH单体)的氢辅助CO活化被认为是Ni(111)表面CO甲烷化的速率决定步骤,其总活化势垒为2.27 eV(图3b)。
尽管CH单体的加氢和最有可能的CH-CH偶联具有相似的激活势垒(0.69 eV对0.71 eV),但由于后一种反应在Ni(111)表面上的吸热反应能更高,因此CH2加氢成CH4比C2H2加氢成C2H4更优0.39 eV(图3b,c)。
与Ni(111)表面相比,TiO2−x/Ni表面的界面位点提高了CO加氢的能量学。例如,在形成CH单体(Ea = 1.03 eV)时,CO活化在TiO2−x/Ni催化剂的界面处增强,这是由于CO/HCO中间体中Ti原子与O原子之间的强吸引相互作用降低了过渡态能量(图3b)。
最有利的CH-CH偶联的激活势垒从Ni(111)上的0.71 eV降低到TiO2−x/Ni上的0.42 eV(图3c),这表明TiO2−x/Ni界面在加速长链烃形成过程中的碳-碳偶联中发挥了重要作用。与直接CH加氢相比,CH-CH偶联在0.17 eV时更容易发生进一步加氢(图3b,c),这主要是由于通过热催化过程,C2产物在TiO2−x/Ni催化剂上具有很高的选择性。
然而,H2O反应物(OH/H)的解离吸附抑制了TiO2−x/Ni催化剂界面处CO的吸附0.24 eV(图3g),导致CH单体的表面覆盖率相对较低,从而导致CH - CH偶联的反应速率较低。然而,这并不能解释TiO2−x/Ni催化剂在光照下C2+具有较高的选择性。在TiO2−x/Ni表面添加一个外部电子来模拟光激发TiO2−x/Ni催化剂,其中TiO2−x中产生的光诱导电子可以从界面Ti转移到Ni(图3a),这一点得到了原位超快红外光谱实验(图2c-f)的证实。
作者发现在光激发的TiO2−x/Ni表面存在一个额外的电子,分别促进了CH-CH偶联和CH直接加氢0.17 eV和0.10 eV(图3d-f)。在光激发的TiO2−x/Ni表面上,较弱的CH中间体吸附(0.39 eV)驱动了较高的放热反应能,从而降低了CH加氢和CH-CH偶联的激活障碍。 CH吸附减弱的原因是光激发的TiO2−x/Ni表面大量电荷转移到CH单体上,并且更多地占据了Ni-C键的反键轨道(图3h,i)。
与TiO2−x/Ni表面相似,在光激发的TiO2−x/Ni表面上,CH-CH耦合仍然比CH直接加氢0.24 eV更有利。额外电子的存在使CO的吸附量略微增加了0.23 eV,这可能是由于CO吸附的光激发TiO2−x/Ni表面通过减小键距(最大为0.06 Å)形成更强的Ti-O键来实现稳定(补充图26)。结果表明:H2O解离吸附能(OH/H)和CO吸附能比较,分别为-2.01 eV和-2.08 eV;图3g)给出了CH-CH偶联CHx中间体相对较高的表面覆盖率,促进了光激发TiO2−x/Ni上C2+的生成。
文献信息
Qin, X., Xu, M., Guan, J. et al. Direct conversion of CO and H2O to hydrocarbons at atmospheric pressure using a TiO2−x/Ni photothermal catalyst. Nat Energy (2024).
