前言
转录组作为研究基因表达的利器,已经成为实验室的常用工具。那么拿到转录组的数据之后,动辄就有几十页的分析报告,几十项分析内容。每个图表的含义是什么,哪一些分析内容是需要重点关注的呢?真正可以用到文章里的分析内容又有哪些?
本次我们对转录组结题报告里的参考基因组比对和数据库注释进行介绍,以方便各位老师快速入手转录组结题报告。
真核转录组参考基因组比对和数据库注释是分析转录组数据的重要步骤,可以帮助识别转录本、注释基因功能和理解生物学过程。下面是这两个步骤的简要介绍:
通常测序生成的reads要与参考基因组或参考转录组进行比对,所以首先需要获取参考基因组和参考转录组信息。
参考基因组比对
参考基因组是指该物种已有的全基因组序列信息以及注释文件,reads比对到参考基因组是数据分析的第一步,后面分析内容都是基于reads比对结果分析的。
比对效率统计
比对效率指Mapped Reads占Clean Reads的百分比,是转录组数据利用率的最直接体现。比对效率除了受数据测序质量影响外,还与指定的参考基因组组装的优劣、参考基因组与测序样品的生物学分类关系远近(亚种)有关。通过比对效率,可以评估所选参考基因组组装是否能满足信息分析的需求。
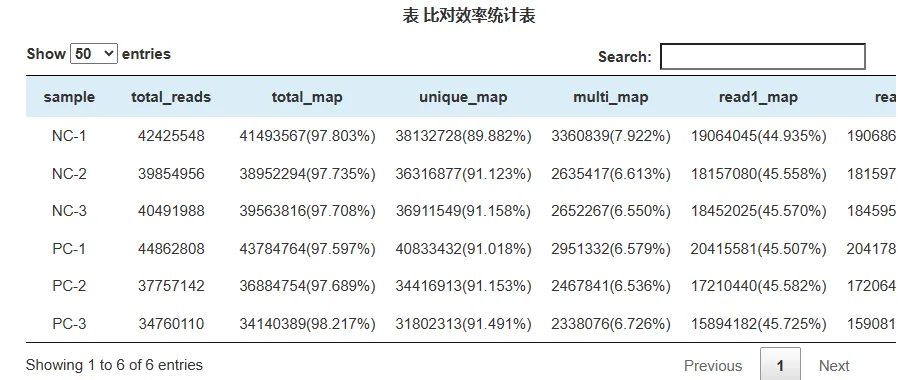
Mapped reads:比对到参考基因组(无参物种是unigenes)的reads,mapped reads并非严格要求100%比对,比对软件一般都会有一定的容错率,一般reads与参考基因组允许最大错配为2个碱基。
Multiple mapped reads:比对到参考基因组多处位置的Reads数目。
Uniq Mapped reads:比对到参考基因组唯一位置的reads。
mapping rate:比对到基因组的reads占clean reads的比值;比对率会随着亲缘关系、基因组组装质量、测序质量、有无污染等有所波动,一般mapping rate大于60%,再低的话就要考虑进行无参组装了。
基因组信息测完之后,接下来就是基因注释,识别这个基因是什么,预测这个基因编码什么蛋白,有什么功能;当获得无参转录组之后需要从头拼接转录本,拼接的转录本功能也需要做注释;当得到了差异表达基因,想做下富集分析,就必须要了解每个基因对应哪个GO分类,也是需要进行功能注释。
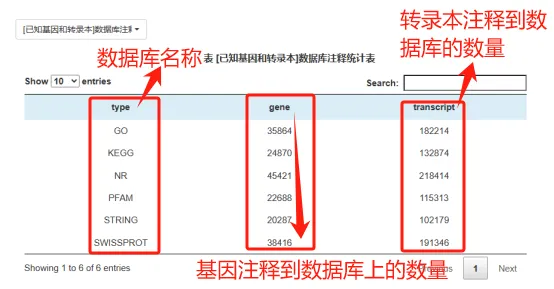
鉴别出新基因(转录本)后,我们会针对所有基因、已知基因、所有转录本和已知转录本四个角度分别进行6大数据库(NR、Swiss-Prot、Pfam、STRING、GO和KEGG)注释,全面获得基因和转录本的功能信息,并对各数据库注释情况进行统计。
一般是对已知基因和转录本的功能注释
对于转录本的基本功能注释通常包括NR、GO、KEGG、COG/KOG、Swwiss-Port。
其中NR和Swiss-Port数据库是两个被广泛使用的蛋白数据库,其中Swiss-Prot是经过严格筛选去冗余的数据库。COG/KOG是基于基因产物的直系同源数据库,而GO和KEGG则是对基因功能和分子通路的数据库。
通过将转录本序列与上述数据库进行比对,就可以得到相对详细的功能注释信息,而后续研究就可以通过注释信息进一步开展。