


概述
近日,南方科技大学王阳刚课题组应美国化学协会Accounts of Chemical Research杂志主编Cynthia J. Burrows教授的邀请,撰写了题为“Modeling Interfacial Dynamics on Single Atom Electrocatalysts: Explicit Solvation and Potential Dependence”的综述论文,系统总结了王阳刚课题组在单原子电催化剂电催化界面动态机制方面的研究进展,并对未来电催化动态模拟领域的研究方向和思路进行了展望。
近十年来,单原子催化剂因其极高的原子利用率和优异的催化活性,在能源电催化领域受到了广泛关注。其中,氮杂石墨烯分散的过渡金属单原子催化剂由于其活性位点结构简单明确,逐渐成为研究单原子电催化剂反应机理和活性趋势的理想模型催化体系。
然而,在实际电化学反应条件下,单原子电催化剂可能会受到复杂电化学界面动力学性质的影响,这使得从简单计算模型中得出的反应活性趋势和机理不一定可靠。本文通过几个典型单原子电催化体系的例子,系统展示了将实际电化学反应条件(如显式溶剂环境、显式电解质离子和电极电位)纳入理论模型,对于正确理解电化学界面物理化学性质、反应机理及其动态机制的重要性。
具体而言,作者分析了实际电化学电位变化对基元电化学反应步骤自由能路径的影响、活性中心电子结构差异对电化学界面溶剂化结构的影响,以及复杂电化学条件对能源电催化反应体系反应动力学和热力学性质的影响。
作者还探讨了复杂电催化界面上的动态机制,如溶剂化效应和界面极化效应所动态调控的反应路径及其稳定的电催化反应中间体、超越催化剂表面的长程动态活性区域,以及单原子位点在实际反应条件下的动态活化或失活机制。基于对单原子电催化体系动态机制的研究,作者进一步指出,传统单原子电催化剂的简单设计原则可能会失效,而将界面丰富的动力学性质考虑在内,则有可能对其进行修订,实现可靠的单原子电催化剂设计。
总而言之,对电化学界面复杂性特别是其动态机制的研究,为电催化理性设计带来了新的机遇,使我们能够更深入地理解和更精细地控制依赖于电位的电化学反应的自由能路径,为进一步设计和优化单原子电催化剂开辟了新的维度。
图文要点
(1)电势依赖的电催化反应自由能路径
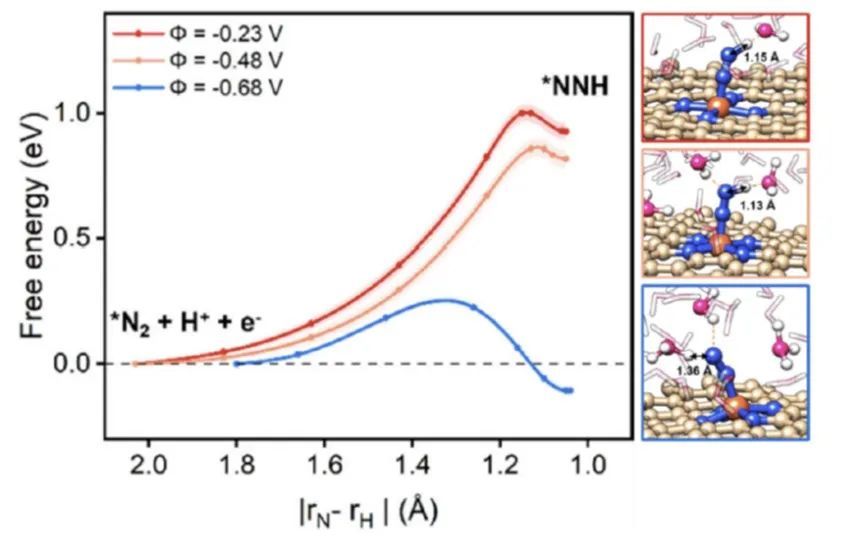
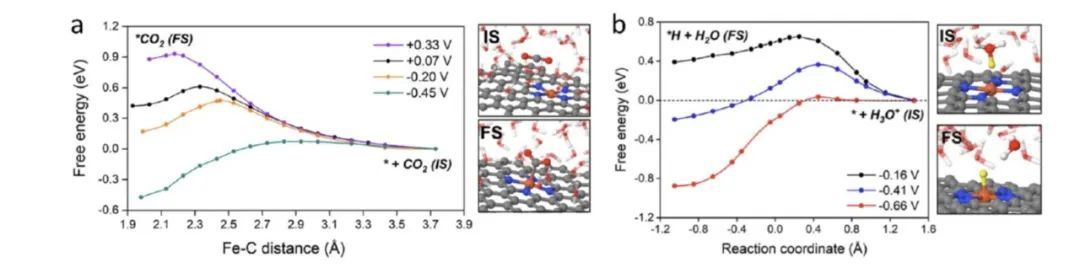
作者最近针对单原子电催化剂电催化NRR,CO2RR,HER等反应的动态模拟显示,电催化反应的路径受到电势的显著影响,特别是过渡态的结构以及在反应坐标下的位置在不同电势下也发生了显著变化,这主要是由于界面溶剂化结构对电势变化的动态响应引起的;当催化活性位点附近的溶剂水重新排列时,它们与反应中间体的高度定向的氢键相互作用也将发生演变,导致反应自由能对电势的依赖性不再像传统计算氢电极模型那样简单。
(2)溶剂调控的单原子电催化反应机理
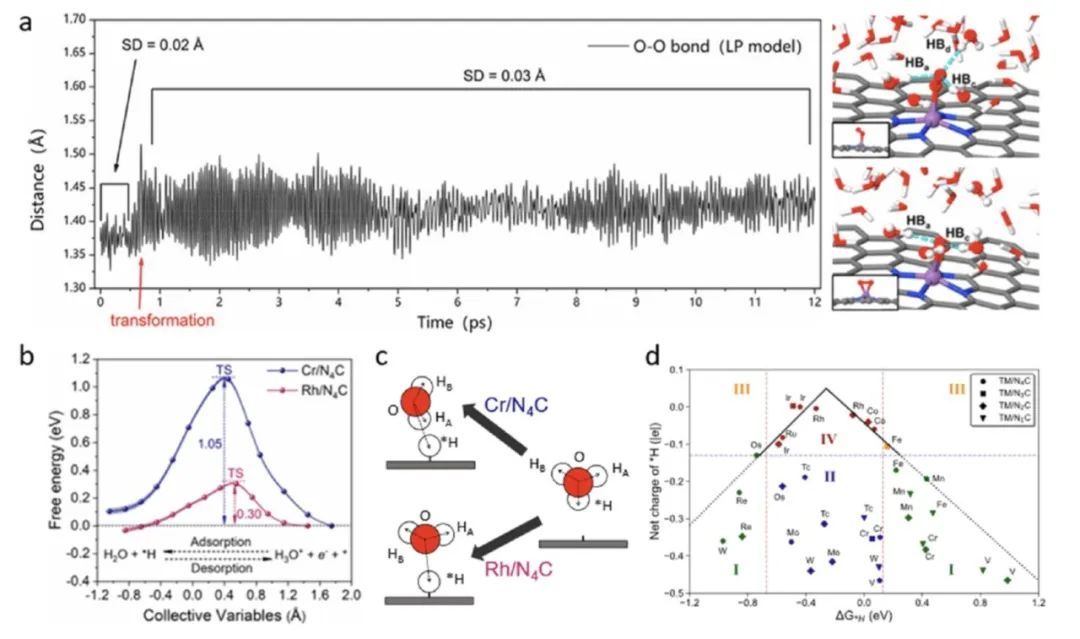
在析氢反应(HER)的研究中,作者发现不同的单原子电催化剂,尽管都具有有利的氢吸附自由能,但其电催化反应的动力学能垒却有可能显著不同,这主要归因于单原子电催化剂界面上水环境与吸附氢中间体之间电荷-偶极相互作用差异。作者对比了H吸附能自由能相近的Cr1N4/C和RhN4/C单原子催化剂,发现二者的反应路径具有明显的差异,在Cr1N4/C单原子催化剂上,氢吸附的终态构型中一个水分子氢端朝下与其发生强烈的电荷-偶极相互作用,导致在吸附过程中水分子需要发生一个强烈的构型重构进而导致该过程具有较高的反应能垒,在RhN4/C单原子上吸附的氢物种上方水的构型则是氧端朝下,吸附过程中不需要经历构型翻转。
催化研究中“火山型曲线”是关联催化反应活性与催化反应中间体结合能的重要曲线,而其之所以成立的本质在于假设了结合能与反应能垒之间一般存在关联,即BEP关系。当BEP关系失效的情况下,“火山型曲线”便不再适用。在针对HER的单原子电催化剂设计中,电催化界面的动力学性质可能起着关键作用,HER过程的能垒不一定与氢吸附自由能直接关联,因此,只依赖于氢吸附自由能(即热力学性质)进行催化剂设计是不够的。
(3)单原子电催化界面的长程动态活性区域

在反应过程中,质子物种(在中性/酸性介质中来源于水合氢离子,在碱性介质则来源于水)可以在体相界面便完成与赝吸附氢氧化物的质子化过程,而无需前往催化剂表面。这将反应区域由单一的催化剂位点扩大到了体相界面区域。这个工作对催化领域的机理研究提供了重要的暗示,即催化反应并不一定严格发生在催化活性位,或许可以通过反应物种/中间体与催化活性位的长程耦合而实现催化过程。
(4)单原子电催化剂的动态活化和失活机制
单原子电催化活性中心可能受到界面动力学性质的强烈影响,并因此而发生动态活化或失活。例如,对于Fe-N4/C催化剂催化ORR而言,当考虑显示溶剂环境的时候,在金属单原子活性中心的背面轴向位置可以与溶剂水分子发生配位,进而改变中心金属单原子的配体场,并因此改变了与氧还原过程中含氧中间体的结合能力,进而改善了氧还原活性;而在Cu-N4/C单原子催化剂电催化CO2还原中,与Cu中心配位的N配体可能在还原条件下发生质子化,进而导致CuN4物种在溶剂环境下被溶解,并成核成Cu团簇,表现出一定的CO2RR活性。
实际电催化反应条件下,许多其他类似的单原子电催化剂可能在不同程度上受到这类动态活化机制的影响(悬挂、溶解、聚集)。通常,当配位原子被质子化时,掺杂的碳载体倾向于削弱金属原子的锚定,这将导致单原子金属中心形成具有更高反应性的热力学亚稳态,但削弱的金属-载体相互作用也有可能导致金属中心从催化剂表面解离而失去活性,一些金属也有可能会重新沉积并聚集成团簇和颗粒,进一步产生电催化活性。

该工作得到了国家自然科学基金优秀青年科学基金项目、面上项目、广东省自然科学基金面上项目以及深圳市科创委基础研究项目等项目的支持。
原文信息及链接
Zhang Z. S.; Li J.; Wang, Y-G.* Modeling Interfacial Dynamics on Single AtomElectrocatalysts: Explicit Solvation and Potential Dependence.Acc. Chem. Res.
