
第一作者(或者共同第一作者): Di Zhang (张頔)
通讯作者(或者共同通讯作者): Li Wei(魏力),Hao Li(李昊)
通讯单位: 日本东北大学,澳大利亚悉尼大学
论文DOI:10.1021/jacs.3c11246
金属-氮-碳(M-N-C)单原子催化剂被认为是高效的铂基催化剂替代品。过去,单原子催化剂在不同pH环境下的活性和选择性机制存在争议。本研究通过分析超过100种M-N-C结构,成功开发了单原子催化剂的pH-电场耦合新模型。该模型揭示了氧还原反应(ORR)活性如何随pH值变化,从碱性溶液的单一活性峰到酸性溶液中的双活性峰,与实验数据高度一致,该成果为开发酸/碱条件下性能优异的非贵金属M-N-C催化剂提供了关键理论机制和设计策略。


封面文章
研究背景
基于金属-氮-碳(M−N−C)组合的单原子催化剂作为氧还原反应(ORR)的催化剂,在近些年获得了极大的关注。这些催化剂因其卓越的ORR催化性能和相对于传统的铂族金属催化剂(PGM)较低的成本,展现出了巨大的潜力。在M−N−C单原子催化剂(SACs)的研究领域内,关于它们的设计、合成、表征、催化性能以及稳定性的评估已经取得了重要的进展。然而,尽管取得了这些成就,科研界仍在探索这些催化剂在不同pH条件下的pH依赖性、对4电子与2电子(4e−/2e−)反应路径的选择性偏好,以及M−N−C SACs在各种pH值条件下的反应速率决定步骤(RDS),目前对这些问题还没有形成共识。
成果简介
本研究深入分析了超过100种M–N–C结构和2000组能量数据,揭示了氧还原反应(ORR)活性火山图随pH变化的特性——从碱性条件下的单一活性峰变化到酸性条件下的双活性峰。研究小组发现,M–N–C催化剂展现出的这种pH依赖性特征,主要是由于它们对氧和过氧氢的吸附体具有适当的偶极矩和极化能力,以及ORR吸附体间的特殊比例关系所致。为了证实理论结果,研究人员合成了一系列具有精确定义的原子配位环境的M–N–C催化剂。实验结果显示,在酸性和碱性环境中的动力学电流密度、塔菲尔斜率和转化率与理论预测高度一致。这项研究不仅提供了对Sabatier原理的新理解,还强调了在设计M–N–C催化剂进行ORR或其他pH敏感电化学反应时,避免所谓的“酸性陷阱”的重要性。

图文导读

本研究首先总结了超过100种M–N–C催化剂在酸性和碱性介质中的实验ORR活性和4e−/2e−选择性,发现基于铁(Fe)的催化剂在不同pH条件下通常展现出较小的依赖性。此外,数据总结发现,即使是以Fe为中心的催化剂,在酸性条件下也表现出不同的ORR性能,这在Fe-吡咯-N4与Fe-吡啶-N4的比较中尤为明显。同时,以钴(Co)为中心的催化剂在酸性条件下通常表现出更高的H2O2选择性,这些观察结果尚未得到现有理论的统一解释。
目前,基于密度泛函理论(DFT)的计算氢电极方法是模拟电催化最常用的方法之一。尽管这种方法能够捕捉到2e−和4e− ORR的趋势,但在缺乏细致的微动力学模型的情况下,预测如电流密度、半波电位和塔菲尔斜率等实验观测结果仍然具有挑战。特别是,由于缺少对pH/电位依赖的自由能的描述,对M–N–C催化剂的pH依赖活性和选择性进行准确建模更加困难。因此,研究突出了开发一个能够准确描述M–N–C催化剂在不同pH条件下ORR行为的统一模型的迫切需求。

在本项研究中,研究团队运用密度泛函理论(DFT)进行计算,深入探讨了金属-氮-碳(M–N–C)单原子催化剂(SACs)的工作机制,特别是在吸附体之间的键合强度比例关系方面的新见解。通过对103种不同中心金属、氮配位数及其周围官能团变化的M–N–C催化剂进行分析,本研究揭示了其吸附特性的差异。与N4配位结构(斜率为1.5,截距为1.26)相比,那些与中心金属原子形成不饱和配位(即配位数为1、2或3的氮原子)的M–N–C催化剂表现出较低的截距(0.7),意味着这些催化剂上ORR相关吸附体的吸附能力更强。这种增强的吸附能力主要归因于金属原子的未饱和氮配位所导致的。同时,较高的截距反映了在特定的HO键合强度下,氧分子的键合相对较弱,这可能会提高O-O键断裂的能量阻碍。研究还发现,O2与HO的比例关系表现为分段线性关系,这在ORR过程中对于HOO*的形成至关重要,为理解4电子向2电子ORR选择性转变的关键点提供了重要前提。
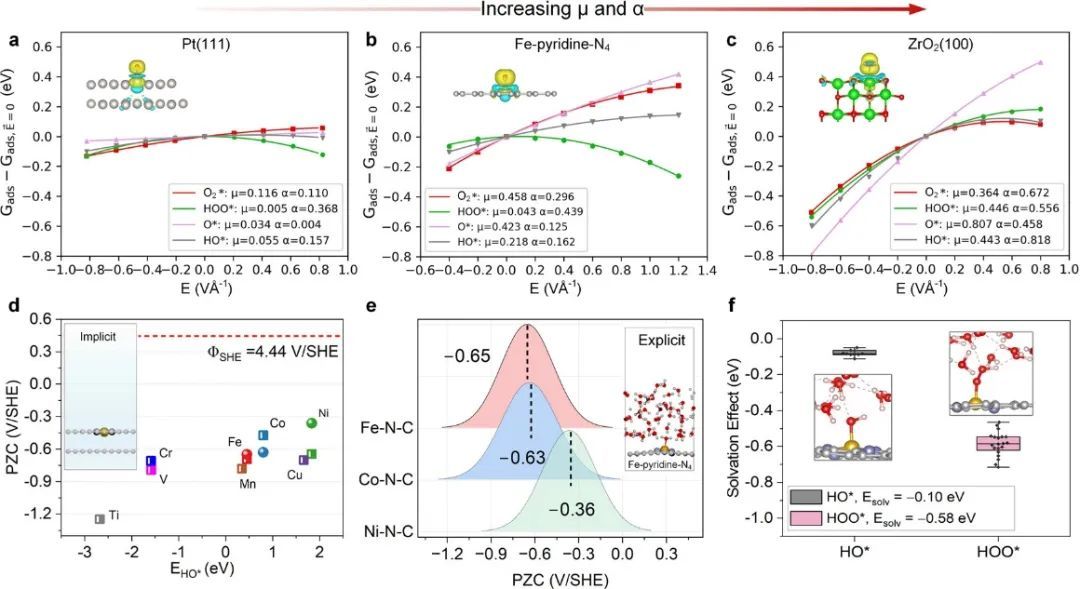
在本研究的框架内,研究团队深入分析了对单原子金属-氮-碳(M–N–C)催化剂氧还原反应(ORR)性能影响显著的各种因素,重点关注了零电荷电位(PZCs)、电场作用以及溶剂化效应。研究中特别考虑了电场如何与那些具有明显偶极矩(μ)和/或极化能(α)的反应中间体及过渡态进行相互作用。通过比较不同环境下的反应性,如上图展示了过渡金属(TMs)、M–N–C催化剂以及过渡金属氧化物(TMXs)上的ORR吸附体对电场变化的敏感度。结果揭示了M–N–C催化剂,尤其是其上吸附的氧(O*)对电场扰动的反应程度,位于过渡金属和过渡金属氧化物之间的中等水平。

进而,本研究展示了金属-氮-碳(M–N–C)单原子催化剂(SACs)在ORR火山图上从碱性环境的单一活性峰向酸性环境的双活性峰的特殊转变。不同于以往的过渡金属或其化合物的ORR活性火山图,M–N–C SACs在0.8 V/RHE下对4e−/2e−反应路径的分离现象,由“酸性陷阱”效应引起。为深化理解,通过对M–N–C催化剂在碱性和酸性条件下4e−和2e−ORR步骤的速率分析,揭示了导致活性峰分离的关键因素。特别地,分析了ORR转化频率(TOF)如何受到pH值变化及特定描述符GHO*的影响。基于以上模型,成功解释了吡啶-N结构在酸性条件下的ORR活性较低,这与实验观察相吻合。M–N–C催化剂中未饱和N配位的独特比例关系降低了约0.5 eV,表明其对于HOO的O-O键裂解的倾向性大于生成H2O2,这一行为降低了H2O2的选择性,也与实验数据分析一致。

为深入探索金属-氮-碳(M–N–C)催化剂的固有活性,研究小组精心设计并合成了13种结构精确的异质分子M–N–C催化剂。这些建构精细的分子催化剂被细分为三个类别:M-COF366、M-Pc以及Fe–Pc–R,依据它们的中心金属元素和相邻的功能团进行分类。通过同步辐射表征技术,上图a-d验证了催化剂中单原子的均一性。接着,通过对比e-h图中的理论计算与实验测定的动力电流密度和塔菲尔斜率,得出i图显示的理论与实验之间的塔菲尔斜率趋势高度一致,从而证实了理论预测在不同pH环境下速率决定步骤的准确性。最关键的是,研究团队通过比较实验观测到的转化率与理论模型预测的转化率,有效地证明了在单原子ORR过程中酸性条件下存在的陷阱现象。
总结展望
在此研究中,团队对M–N–C催化剂在氧还原反应(ORR)中的pH敏感性进行了深入探讨。这些催化剂对吸附体如氧(O)和过氧氢的特定偶极矩和极化特性,以及ORR相关吸附体之间的比例关系,是其pH依赖性的关键。研究涵盖了影响2电子和4电子ORR路径的各种因素,包括热力学性质、动力学特性、零电荷电位(PZCs)和环境效应的综合考量,从而揭示了酸性和碱性条件下不同的速率决定步骤(RDS)。根据经典的Sabatier原则,有效的催化剂需要实现对吸附体的适度结合强度,既不过强也不过弱。然而,研究表明,在酸性条件下,避免将M–N–C催化剂设计成落入所谓的“酸性陷阱”是至关重要的,该条件下无论是2电子还是4电子的ORR反应效率都会显著降低。基于这些见解,研究提出了一个更为精确的理论模型,预测M–N–C催化剂在不同pH条件下的活性和选择性。
课题组介绍
日本东北大学材料科学高等研究所 HaoLi Lab:
课题组负责人:李昊,副教授,2022年起任职于日本东北大学(Tohoku University)材料科学高等研究所(AIMR),作为课题组负责人从事材料设计与计算、人工智能(机器学习和数据科学)开发研究。2019年博士毕业于美国德克萨斯大学奥斯汀分校化学系及Oden科学与工程计算中心(师从反应过渡态算法创始人之一Graeme Henkelman教授)。2017年在加州大学洛杉矶分校的纯粹与应用数学中心(IPAM)担任访问研究员。2020-2022年初于丹麦科技大学物理系从事博士后工作,师从现代催化理论鼻祖、美国工程院院士Jens K Nørskov教授(美国工程院院士、丹麦双院院士、欧盟院士)。2014年至今已发表论文181篇,包含Nature Catalysis、Nature Communications、Journal of the American Chemical Society、Advanced Materials、ACS Catalysis等领域权威杂志。总引用>7500余次、h指数为49。 第一作者:张頔,日本东北大学特聘助理教授,2021年博士毕业于上海交通大学,主要研究方向为材料计算理论与人工智能方法开发,电催化过程理论建模与预测,先进氢能源材料设计与开发。目前,以第一作者在Nature Communications, JACS, Advanced Materials等国际知名期刊发表论文。
澳大利亚悉尼大学魏力课题组
课题组负责人:魏力,2011年博士毕业于新加坡南洋理工大学,现任澳大利亚悉尼大学副教授,澳大利亚研究理事会(ARC)未来研究员,研究方向为高性能电催化剂开发,开发纳米尺度上实现对材料组装控制的新方法。发表综述、期刊论文130余篇,书籍章节1篇,会议论文2篇,会议论文33篇摘要,总引用11664次,h-index 52。
