

成果简介
强金属-载体相互作用(Strong metal-support interaction, SMSI)因其在稳定活性金属和调节催化性能方面的重要作用而在多相催化中得到了广泛的研究,但SMSI的起源尚未完全揭示。
近日,北京化工大学卫敏教授、杨宇森副教授、北京大学马丁教授、北京科技大学邢献然教授等人通过使用Pt/CeO2作为模型催化剂,报道了在Pt和CeO2(110)面界面处的嵌入结构,其中Pt簇(~1.6 nm)嵌入到3-4原子层的CeO2晶格中。相反,这种现象在CeO2(100)载体中不存在。这种独特的几何结构作为一种有效的诱导剂,引发了更多的电子从Pt团簇转移到CeO2(110)载体上,并形成了界面结构(Ptδ+-Ov-Ce3+),这对稳定Pt纳米团簇起着至关重要的作用。
基于实验研究和理论计算的综合研究表明,界面位点是水气转换反应(WGSR)的本征活性中心,具有中等强度的CO活化吸附和大幅降低的H2O解离能垒,这是Pt/CeO2(110)具有突出的催化活性的原因(250℃时反应速率为15.76 molCOgPt-1h-1,转化频率为2.19 s-1)。
此外,Pt/CeO2(110)催化剂在连续工作120 h的测试中表现出优异的耐久性,远远优于Pt/CeO2(100)催化剂,这证明了这种嵌入结构在提高催化剂稳定性方面的优势。 相关工作以《Highly Stable Pt/CeO2Catalyst with Embedding Structure toward Water−Gas Shift Reaction》为题在《Journal of the American Chemical Society》上发表论文。
图文导读

其TEM和HRTEM图像显示CeO2纳米棒和纳米立方体的结构特征明显,主要暴露(110)和(100)面。在以0.6 wt %的负载量锚定金属Pt后,这两种Pt/CeO2样品的PXRD图显示CeO2相的面心立方(fcc)结构,没有观察到Pt相(图2a), Pt物种高度分散。经AC-HAADF-STEM图像显示,这两种Pt/CeO2样品的CeO2载体上的超小Pt纳米团簇(平均尺寸:1.6-1.9 nm)占主导地位(图1a-i)。
值得注意的是,AC-HAADF-STEM图像和能量色散光谱(EDS)揭示了Pt/CeO2(110)样品的一个有趣的界面结构,其中Pt原子进入载体并占据3-4原子层内的CeO2晶格,形成深嵌入界面结构(图1a-c)。在Pt/CeO2(110)样品中还观察到另一个有趣的现象,其中Pt簇作为一个整体嵌入到几个原子层中的CeO2中(图1e-g)。相比之下,对于Pt/CeO2(100)样品,Pt簇仅位于载体表面,而没有发生Pt原子嵌入(图1i-k)。

此外,SR-PXRD(图2b)以及精修结果表明,Pt/CeO2(110)样品中Pt嵌入到载体中,在Pt负载后CeO2衍射角度略有下降,晶格参数从5.412增加到5.418 Å(晶格膨胀)。然而,Pt/CeO2(100)的晶格参数变化可以忽略不计(图2b)。这与AC-HAADF-STEM观测到的Pt(111)晶格间距与CeO2(110)面的匹配度优于与CeO2(100)面的匹配度(晶格间距差距:0.352 vs 0.441 Å)有关。
根据以上结果,两种Pt/CeO2样品的界面原子结构示意图模型如图1d、h、l所示,表明Pt/CeO2(110)体系比Pt/CeO2(100)体系具有更强的金属-载体键合强度。此外,还在Ir/CeO2(110)和Au/CeO2(110)样品中发现了类似的部分嵌入结构,其中Ir或Au簇作为一个整体嵌入到CeO2载体中。这种独特的几何结构将诱发强烈的金属-载体相互作用,这可能对电子结构(例如,电子转移)和金属纳米团簇的稳定性(例如,抗烧结和聚集性)产生影响。
通过H2程序升温还原(H2-TPR)、原位CO-DRIFTS和XANES研究了Pt团簇与不同CeO2表面的相互作用强度。对这两种样品的还原过程进行H2-TPR研究(图2c),观察到两个低温峰和一个高温峰,分别归属于Pt物种(α)、界面氧物种(Pt-O-Ce)(β)和表面晶格氧CeO2(γ)。从Pt/CeO2(100)到Pt/CeO2(110),β峰向更高的温度移动,表明后一种情况下金属-载体相互作用更强。
通常情况下,金属-载体的相互作用伴随着界面处的电荷转移,这导致金属的电子密度发生变化,从而导致CO探针分子的化学吸附强度发生变化。如图2b所示,CO在这两种Pt/CeO2样品上的原位化学吸附,其中高波数峰和低波数峰分别归因于CO在带正电的Pt物种(Ptδ+)和Pt0位点上的线性吸附。
在高波数峰,观察到从Pt/CeO2(100)到Pt/CeO2(110)中的7 cm-1的蓝移,表明Pt/CeO2(110)样品中从Pt到载体的电子转移更强。这导致铂的电子密度降低,从而减轻了CO的吸附,导致v(CO)发生蓝移。因此,Pt/CeO2(110)中存在较高的Pt氧化态,这与铂-铈界面紧密的嵌入结构有关。 Pt L3边缘XANES谱图进一步揭示了电子相互作用(图2e)。
值得注意的是,由于Pt物种的部分氧化,两种样品都表现出相对于Pt箔(Pt0)的白线强度增加,但Pt/CeO2(110)样品的白线强度增强更明显,这与H2-TPR和原位CO-DRIFTS的结果一致。在EXAFS光谱(图2f)中可以看出,Pt/CeO2(110)中的Pt-Pt键比Pt/CeO2(100)和Pt箔中的Pt-Pt键要短得多,这是由于Pt-CeO2之间的键合作用更强。
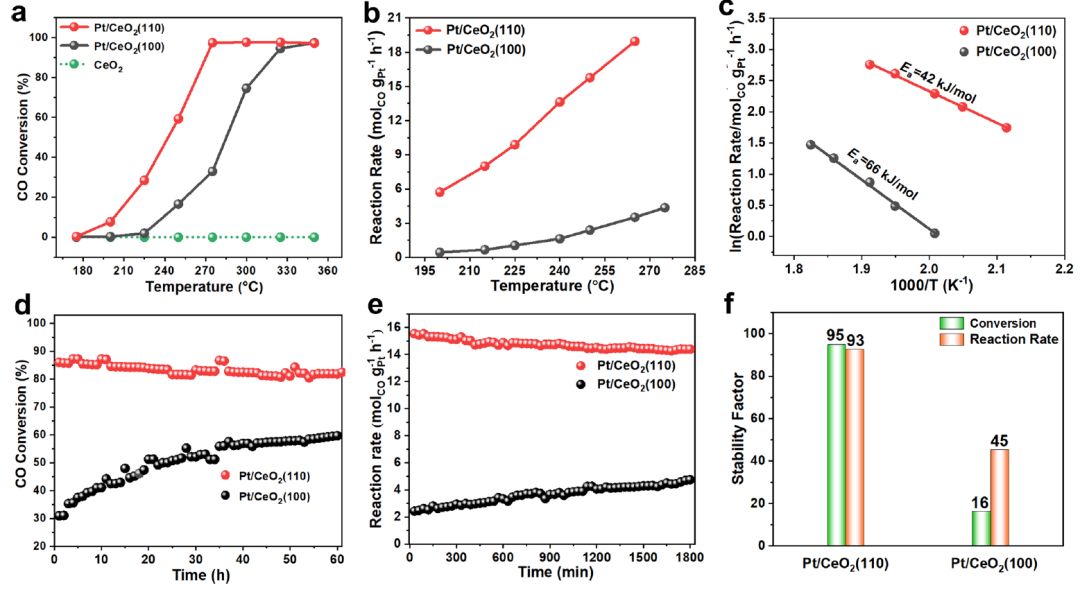
在175~350℃的温度范围内,对两种Pt/CeO2样品对WGSR的催化性能进行了评价。两者对CO2均有绝对选择性(100%),表明不存在甲烷化副反应。然而,与Pt/CeO2(100)样品相比,Pt/CeO2(110)样品表现出更高的催化活性(图3a、b),在250°C下的反应速率为15.76 molCOgPt-1h-1,TOF值高达2.19 s-1,优于目前最先进的催化剂。相应地,表观活化能(Ea)给出如下顺序(图3c):Pt/CeO2(110)<Pt/CeO2(100)。
图3d显示了两种Pt/CeO2催化剂在275℃下的长期稳定性。对于Pt/CeO2(110)催化剂,在60 h的测试中,CO转化率略有下降(从86%降至82%)。值得注意的是,对于Pt/CeO2(100),在前40小时内观察到CO转化率的增强(从30%到57%),然后在接下来的20小时内达到稳定状态(~59%),但与Pt/CeO2(110)相比,它仍然处于相对较低的水平。
图3e显示了反应速率与反应时间的关系,这是在250℃下CO转化率较低(<15%)的情况下进行的,以排除扩散的影响。对于Pt/CeO2(110),反应速率从15.54 molCOgPt-1h-1稍降低到14.40 molCOgPt-1h-1;而对于Pt/CeO2(100),反应速率从2.42增加到4.75 molCOgPt-1h-1,对应活化诱导性能增强。稳定性因子被定义为稳定性测量期间CO转化率(或反应速率)变化的倒数,被用作表示催化剂稳定性的标准化描述符。如图3f所示,稳定性系数更高的Pt/CeO2(110)在CO转化率和反应速率方面都优于Pt/CeO2(100)。

本文利用XPS进一步研究了铂-铈相互作用对电子结构的影响。这些测试样品的Pt 4f7/2轨道显示出两个峰,分别位于~71.3和~72.4 eV,归属于Pt0和Ptδ+物种(图4a),这表明电子从Pt转移到界面处的载体。 本研究利用原位DRIFTS结合原位拉曼光谱研究了WGSR的内在活性位点。
如图4b、c所示,在H2/He气氛中350℃预还原后,将得到的Pt/CeO2催化剂先暴露于CO气氛中,再暴露于250℃的H2O蒸气中进行信号采集。在~2082和~2063 cm-1处观察到两个谱峰,分别归因于CO在Ptδ+和Pt0位点的化学吸附。随着水蒸气的引入,CO在Ptδ+处的化学吸附逐渐减少,在2400~2300 cm-1处出现了一个新的峰,该带属于CO2分子中CO的伸缩振动,而Pt0处的CO化学吸附仍然存在。随后,2400~2300 cm-1处的峰逐渐减弱,表明CO2从催化剂表面解吸。这一现象证明,界面Ptδ+是CO活化的活性位点。
此外,在250°C下进行H2O蒸汽和CO气氛的原位循环脉冲(图4d)。将CO气体脉冲进入反应池后,只得到少量的CO2;随后的水蒸汽脉冲立即导致H2和CO2的大量产生,这与原位DRIFTS的结果一致。
本文强调了可还原载体上的氧空位在含氧物质的活化吸附中起着至关重要的作用,并利用原位拉曼监测了氧空位浓度的变化。缺陷诱导模式(D)峰(~590 cm-1)与八面体局部对称振动模式(F2g)峰(~460 cm-1)之间的相对强度比(D/F2g)表示氧空位的相对浓度,如图4e所示。收集了两种Pt/CeO2催化剂在不同气氛下的原位拉曼光谱(图4e)。原位预还原后,发现Pt/CeO2(110)比Pt/CeO2(100)表现出相对较高的氧空位浓度(0.487 vs 0.246),这与XPS结果一致。
随后,随着250℃下H2O蒸气的引入,两种样品中的D/F2g值降至较低水平(~0.10),表明H2O分子在氧空位处进行了活化吸附,导致氧空位减少。

众所周知,CO在Pt表面的强化学吸附通常会导致催化剂中毒,这是Pt基催化剂难以获得高催化活性的原因。为了评价反应物对反应的影响,测量了CO和H2O的反应级数(图5a、c)。Pt/CeO2催化剂均呈现负的CO反应级数(图5a), Pt/CeO2(100)的CO反应级数高于Pt/CeO2(110),表明CO分子对WGSR有抑制作用。在Pt/CeO2(100)中较明显的抑制作用主要来源于Pt表面对CO的极强吸附,而在Pt/CeO2(110)中,由于电子密度较低的Ptδ+物种使CO的化学吸附减弱,这种抑制作用得到了有效的缓解。
DFT计算表明,CO在Pt/CeO2(100)和纯Pt(111)上的吸附能大于Pt/CeO2(110)上的吸附能(-1.76/-2.02 vs -1.25 eV;图5 b)。此外,采用CO氧化过程作为探针反应,测试了两种Pt/CeO2样品的催化性能。在具有嵌入结构的Pt/CeO2(110)中,缺乏电子的Pt簇上CO结合减弱,导致CO氧化活性较高,而Pt/CeO2(100)表现出较差的行为。
在Pt/CeO2(110)催化剂上,H2O的反应级数为0.26(图5c),低于Pt/CeO2(100)的反应级数0.66,说明在Pt/CeO2(100)催化剂上,水解离对反应速率的影响不显著。此外,Pt/CeO2(110)在界面氧空位处的H2O吸附能大于Pt/CeO2(100)(-0.60 vs -0.35 eV)(图5d),但解离能垒(0.37 eV)远低于后者。然而,在Pt/CeO2(110)和Pt/CeO2(100)的非界面氧空位处,H2O的解离更加困难。这证实Pt/CeO2(110)中Ov-Ce3+的界面位点是更有利于水分子解离吸附的活性中心,符合水分子的较低动力学反应级数。
因此,实验研究结合理论计算证实,在Pt/CeO2(110)中,嵌入界面结构诱导的具有更强电荷转移和更高氧空位浓度的界面位(Ptδ+-Ov-Ce3+)优化了CO的吸附,加速了H2O的解离,从而具有非凡的催化活性。
在反应条件(CO+H2O,275°C)下,对Pt L3边缘进行了原位EXAFS,以监测两种Pt/CeO2样品配位环境的细微变化。对于Pt/CeO2(110)催化剂,除了Pt-Pt和Pt-O配位外,还检测到配位数为~0.7的Pt-Ce壳层(图5e),证实了Pt和Ce之间直接接触的紧密嵌入界面结构。稳定性测试后,通过AC-HAADF-STEM和EDS图谱没有观察到Pt簇尺寸的明显变化(从1.63 nm到1.66 nm)。
文献信息
Highly Stable Pt/CeO2Catalyst with Embedding Structure toward Water–Gas Shift Reaction,Journal of the American Chemical Society,2023.
