
1. Nat. Commun.: 差分相衬(DPC)电子显微镜直接观察缺陷电场中的原子级极化

电催化水分解产氢是缓解全球化石燃料危机的有效方法。这个过程涉及相间反应,需要开发先进的电催化剂来促进反应的进行。随着纳米、亚纳米和原子尺度材料设计的深入研究,在原子水平上揭示电催化性能起源对于开发高效、低成本的催化剂至关重要。事实上,电催化过程的本质是反应物和催化剂之间的电荷转移,从而使得反应物的吸附和它们的连续活化/转化。电催化剂的电荷转移效率取决于这些表面原子位点周围的本征场/电荷分布,许多先进的电催化剂表面具有原子缺陷结构,这些缺陷可以改变电催化剂的电场/电荷分布,提高催化性能。
更重要的是,在电催化剂表面引入原子缺陷导致产生非周期性电场,其可以显着增强反应界面处的电场效应,有助于调节催化反应动力学。因此,对电催化剂上的非周期电场进行高精度的空间分析是理解催化机理的关键。但是,目前原子成像技术很难对特定原子缺陷周围的非周期性电场进行表征,阻碍了含缺陷催化剂的原子电场与催化活性之间的结构-性能关系研究。


近日,浙江大学陆俊、电子科技大学罗俊、温州大学袁一斐和郑州大学邵功磊等以富含原子缺陷的单层二硫化钼(Mo2S)为模型催化剂,结合先进的电子显微镜和差分相衬(DPC)技术,观察了反位缺陷周围极化电场的分布。
具体而言,研究人员首先通过化学气相沉积和煅烧法制备了具有Mo空位(VMo-MoS2-1)和反位缺陷(S2Mo-MoS2-5)的MoS2,并测试了它们的电催化HER活性。结果表明,在酸性条件下,具有反位缺陷结构的S2Mo-MoS2-5催化剂在10 mA cm−2电流密度下的过电位仅为169 mV,Tafel斜率为56 mV dec−1,并且该催化剂还表现出优异的稳定性。

密度泛函理论(DFT)计算表明,MoS2中Mo空位的引入导致产生具有两个Mo-S键的双配位S原子,其不饱和性质产生相对更强的H*吸附;而S2Mo-MoS2催化剂中的双配位S原子表现出更明显的结构扭曲,这将削弱H*的吸附。此外,对于具有两个Mo-S键和一个S-S键的三配位S原子,比Mo-S键更弱的S-S键使得H*吸附强于MoS2;相反,额外的S-S键导致H*吸附比VMo-MoS2中二配位S原子更弱。同时,研究人员利用DPC技术观察了反位缺陷周围极化电场的分布。
结果显示,反位缺陷结构的电荷密度分布是不对称的,这也与DFT计算一致,进一步证明反位缺陷原子电场的极化直接导致了不对称电荷分布的出现,增强了H*的吸附,优化了催化活性。总体而言,反位缺陷的形成打破了活性S原子的原有配位状态,引入了更有利于H*吸附的新配位环境,从而提高了催化活性。总的来说,该项工作不仅揭示了原子级缺陷对催化活性的影响,而且为利用微机电系统(MEMS)装置在显微镜下实现实时催化研究奠定了基础。
Atomic-level polarization in electric fields of defects for electrocatalysis. Nature Communications, 2023. DOI: 10.1038/s41467-023-43689-y
2. Nat. Commun.: 构建CoP/rGO@Ti催化剂,实现高效耐盐电催化海水分解

氢能作为一种高热值的可再生清洁能源,被认为是替代传统化石燃料的最理想的能源之一。其中,电化学水分解是最有前景的制氢技术。目前,工业电催化制氢仍以高纯水为原料,而淡水资源的稀缺和净化工艺的高成本限制了电化学水分解技术的商业化应用。由于世界上97%的水资源都存在于海洋中,电催化海水分解被认为是其他制氢方法的合适替代品。
然而,由于海水化学成分复杂,大量溶解的离子盐(尤其是氯离子),容易使催化剂中毒,降低海水分解的耐久性。此外,在海水分解过程中,随着盐浓度的增加,催化剂的催化活性降低得更明显。因此,开发高效、稳定、耐盐、耐腐蚀的电催化剂是推进海水电解工业应用的关键。


近日,西北工业大学贺亦柏、史俊清和中国科学院赣江创新研究院黄小磊等通过一种简单的制备策略设计并构建了一种高效的无粘结剂CoP/rGO@Ti (CoPGT)海水分解电极电催化剂。
具体而言,研究人员首先通过分子动力学模拟预测分子和离子在CoP催化剂表面的分布情况。在催化剂上施加偏压时,盐离子被排出到水层外,而水分子优先吸附在CoP催化剂表面的几个原子区域,形成一个薄水层;由于这种行为减少了催化剂表面离子的竞争,CoP催化剂在海水分解过程中对电解质中盐的积累具有明显的抗性,从而获得了良好的电催化性能。
值得注意的是,作为对比样的Co3O4催化剂表面的各种离子是随机分布的,没有形成明显的水层,大量的离子吸附在催化剂表面,与表面的水分子竞争活性中心,阻碍了电解水反应的进行。


电化学性能测试结果显示,所制备的CoPGT催化剂具有良好的结构稳定性和较高的催化活性,在电流密度为10 mA cm−2时,过电位仅为103 mV,并在碱性介质中具有稳定的长期性能;即使在电流密度高达200 mA cm−2时,该催化剂的过电位低至210 mV,远低于20% Pt/C催化剂(300 mV)。
更重要的是,由于CoP对Cl−具有有效的排斥作用,随着盐浓度的增加,CoPGT仍能保持良好的催化性能和稳定性(在10 mA cm−2电流密度下,Cl−浓度从0 M到饱和,过电位增加<28 mV);此外,与20% Pt/C催化剂(溶解度为2.37%)相比,CoPGT催化剂在海水分解过程中的溶解度仅为0.04%,表现出更好的耐腐蚀性能,这与模拟结果一致。
Corrosion-resistant cobalt phosphide electrocatalysts for salinity tolerance hydrogen evolution. Nature Communications, 2023. DOI: 10.1038/s41467-023-43459-w
3. Nat. Commun.: SSZ-13沸石中引入Cu-OH单位点,促进甲烷直接氧化制甲醇

甲烷(CH4)作为全球储量丰富的能源,寻求更有效、更经济的利用方式已引起人们的广泛关注。甲烷直接氧化转化为甲醇(CH3OH)(MTM),在多相催化中是一个很有前途的反应,它提供了在温和的操作条件下将甲烷转化为高价值化学品和可凝聚能源载体的潜力。
然而,由于甲烷C-H键的高度稳定性(解离能为435 kJ mol−1)以及热力学上有利的甲烷过氧化,使得甲烷直接、选择性地转化为甲醇变得极其困难。为了实现有效的CH4催化转化,关键是要通过合成一种合适的催化剂,以减少C-H键断裂的活化能,并提供CH3OH的高选择性。
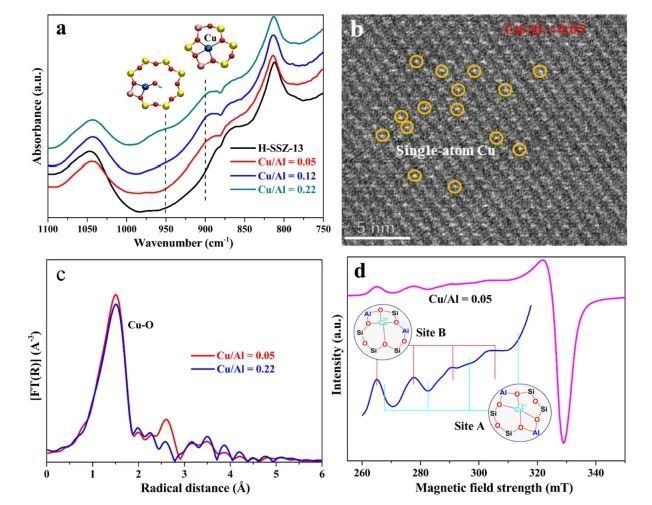

近日,华盛顿州立大学王勇、厦门大学熊海峰和王帅等采用离子交换法在CHA分子筛的六元环(6MR)空隙中引入单Cu-OH位点(Cu1/SSZ-13),并将其作为催化甲烷直接氧化转化的催化剂。
实验结果表明,Cu1/SSZ-13催化剂在400 ℃下的CH3OH时空产率(STY)和选择性分别高达2678 mmolCu−1 h−1(218 μmolgcat.−1 h−1)和93%;同时,这种限制在小6MR空隙中的单个Cu活性位点也可用于好氧的MTM反应,在较高的反应温度(>350 ℃)下也实现了高的CH3OH产率和选择性,以及具有低的CO2(过氧化产物)产量。

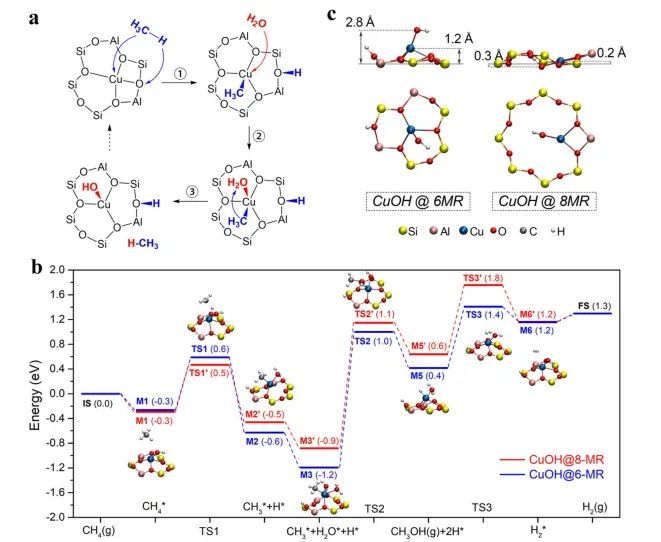

理论计算表明,与具有较大空隙的8MR相比,6MR空隙的高催化效率是由于其具有更强的空间限域效应,其作为Cu-OH单位点的载体能够有效改善CH4到CH3OH的转化;并且6MR中裸露的Cu(II)单原子位点在甲烷C-H活化中具有活性,但其稳定的四配位结构阻碍了它们在较低温度(≤200 °C)下的反应活性。
此外,基于原位光谱、同位素标记和密度泛函理论计算,提出了6mr 受限 Cu (II)单位上甲烷转化为甲醇的可能反应路径:CH4上的H通过与Cu-OH的O原子结合而活化C-H,导致Cu-OH演变为HOH*-Cu-CH3*结构,随后H2O在Cu原子上吸附产生HOH*-Cu(H2O)-CH3*过渡态;该过渡态上形成CH3OH分子并发生解吸转化为HOH*-Cu-H*物种,最后来自Cu-H*和HO-H*的H*原子组合产生H2,伴随着Cu-OH活性位点的再生。
综上,该项研究为深入理解Cu沸石催化剂在甲烷水蒸气连续转化制甲醇反应中的应用提供了有价值的见解,并为该领域的进一步研究奠定了基础。
Confined Cu-OH single sites in SSZ-13 zeolite for the direct oxidation of methane to methanol. Nature Communications, 2023. DOI: 10.1038/s41467-023-43508-4
4. JACS: 调控Pd纳米材料的形貌,显著促进乙炔半加氢反应

金属纳米催化剂的原子表面结构对催化过程中的化学活性和选择性的调节具有决定性作用。归因于纳米催化剂三维结构的局部性,这挑战了传统方法的空间检测极限,但为揭示内在催化机理奠定了基础。在纳米催化剂表面周围,有许多重要的结构特征,包括表面的配位缺陷、晶格应变和梯度分布以及对称性的演化和破坏。
实际上,改变相邻的配位环境,可以在纳米催化剂表面提供不同的吸附形式、负载能力和迁移能力。此外,调整活性位点之间的原子间距也会驱动化学键的转换,从而控制断裂键的重新形成和反应物质的产生。然而,从实验结果中对纳米催化剂的原子尺度的三维结构分析仍存在困难,这限制了在原子水平上揭示催化反应机理和开发高性能催化剂。
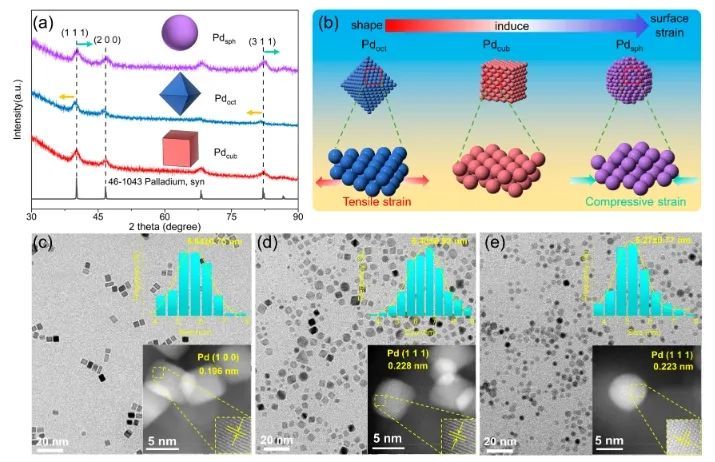
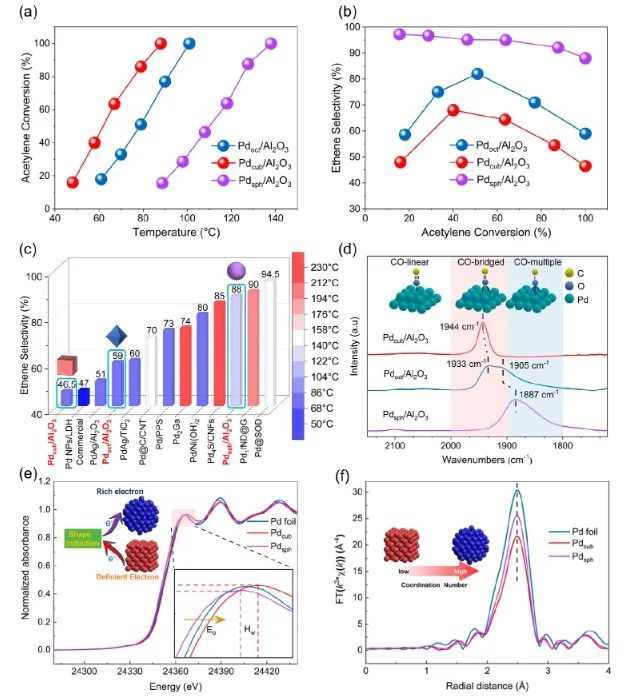
近日,北京科技大学邢献然和李强等对Pd纳米催化剂进行了原子水平的三维深入研究,并用对分布函数(PDF)验证了其形状-结构-吸附关系。研究人员以Pd催化乙炔半加氢为例,通过纳米催化剂的形貌调控(立方体、准八面体和球体),Pd纳米球催化剂的乙炔加氢选择性提高了88%,远高于商业PdAg/Al2O3催化剂(51%);更重要的是,在反应过程中,Pd球的乙烯选择性达88%,高于Pd立方体和Pd准八面体,甚至超过了已报道的单金属Pd纳米催化剂。
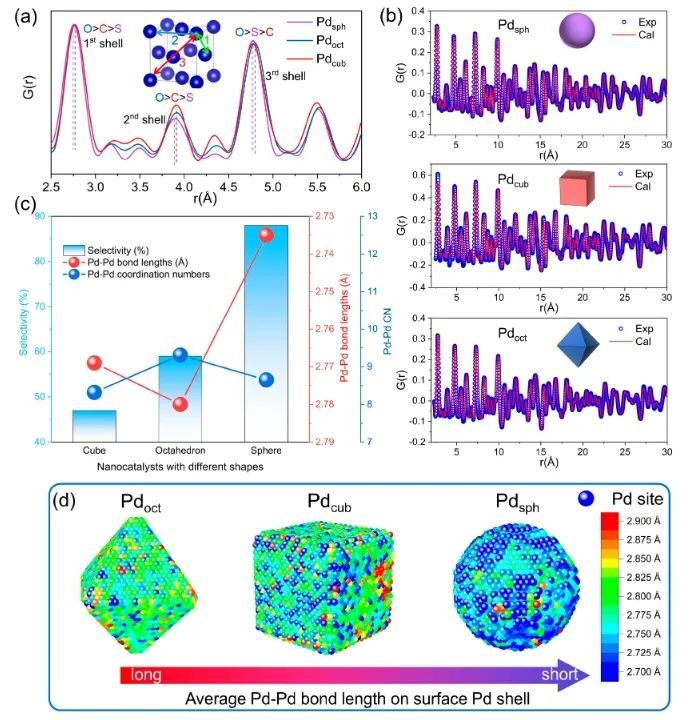
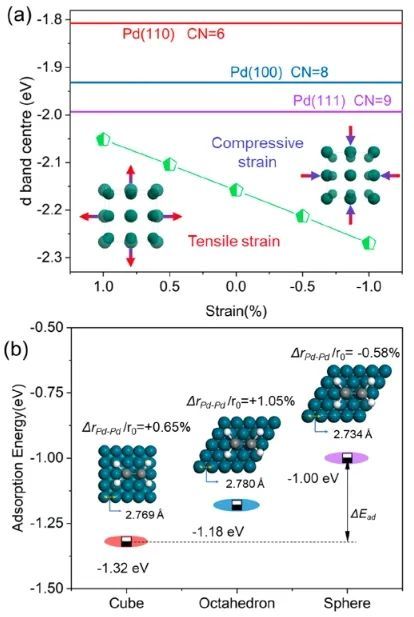
原子对分布函数(PDF)和逆蒙特卡罗模拟(RMC)表明,与立方体状和准八面体状Pd纳米催化剂相比,Pd球纳米催化剂表面存在更多相邻的Pd-Pd对和异常的压缩应变,这导致催化剂的d带中心下移,缓解了反应中间体的吸附,从而提高了反应活性和乙炔半加氢生成乙烯的选择性。总的来说,该项工作揭示了三维结构的调控对催化剂性能的影响,这可能为设计高效的纳米催化剂和深入了解表面结构和化学活性之间的关系提供指导。
Atomic three-dimensional investigations of Pd nanocatalysts for acetylene semi-hydrogenation. Journal of the American Chemical Society, 2023. DOI: 10.1021/jacs.3c08619
5. Angew: 聚合物调节反应微环境,增强CO2电还原制C2H4

化石燃料的短缺和二氧化碳(CO2)的过度释放严重影响了人类的活动和污染了生态环境。为了应对这些挑战,将CO2转化为增值化学品对于建立循环经济至关重要。电催化CO2还原(CO2RR)是一种缓解能源和环境问题有前景的方法。在众多金属催化剂中,Cu由于其合适的*CO偶联强度和对*H中间体的弱吸附能力,被认为是CO2RR产生C2+产物的有效催化剂。然而,CO2RR过程中涉及的多步质子转移往往导致对特定产物的选择性和能量转换效率较低。因此,寻找高选择性和高效的催化剂,以将CO2还原为高附加值的产品是推动CO2RR实际应用的关键。

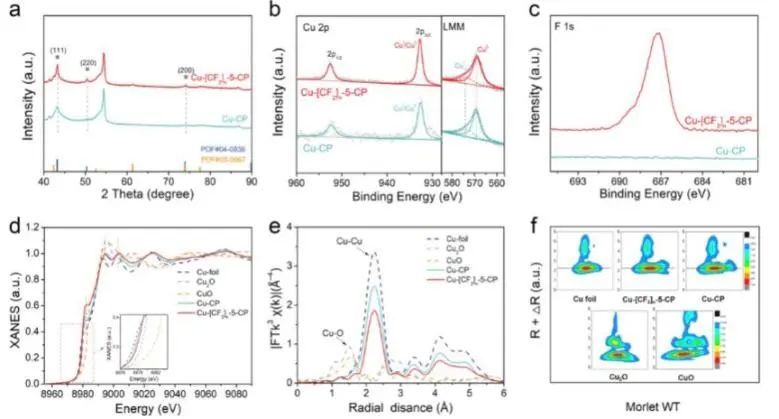
近日,中国科学院化学研究所韩布兴、华东师范大学吴海虹和贾帅强等报道了一种简便的聚四氟乙烯(PTFE)处理策略,合成了一种高效的Cu基CO2RR电催化剂(Cu-[CF2]n-x-CP)。实验结果表明,在−1.25 VRHE下,最优的Cu-[CF2]5-x-CP催化剂上CO2电还原为C2H4的法拉第效率(FE)约为67.25%,C2H4部分电流密度为36.66 mA cm−2,优于Cu-CP和Cu/PTFE-CP电极。同时,该催化剂还具有优异的稳定性,其在连续11小时CO2电解过程中FEC2H4和电流密度几乎未发生下降,且反应后材料的结构也保持良好。

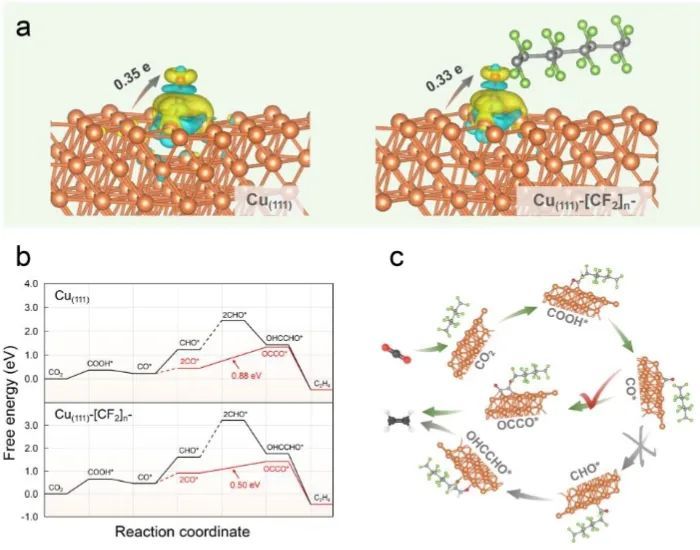
光谱表征和理论计算表明,改性的Cu基电极具有超嗜气性和超疏水性,从而构建了具有CO2气泡/液体电解质/电极的“气-液-固体”微环境。该微环境有利于气态CO2的捕获,延长其在电极表面的停留时间。同时,疏水多孔的Cu-[CF2]n-5-CP仅部分接触电解质,增强了CO2的扩散和吸附,增强了活性位点的可及性;并且该催化剂为CO2RR创建了丰富的三相结,促进了CO2的快速转化。
此外,表面润湿性工程可以调节电极表面附近的质子和CO2的浓度,电极表面的疏水层抑制了水分子的扩散,导致电极表面的OH−浓度增加,而较高的表面pH值有利于C-C耦合,从而促进了C2H4的生成。总的来说,该项工作证明了原位合成策略在改性CO2电催化剂反应界面微环境方面的可行性,相信这种策略在金属基催化剂的设计和商业化以及乙烯的商业生产方面具有更大的实用性和多功能性。
Polymer modification strategy to modulate reaction microenvironment for enhanced CO2 electroreduction to ethylene. Angewandte Chemie International Edition, 2023. DOI: 10.1002/anie.202313796
6. Angew: Bi2Sn2O7中引入亚纳米Bi团簇,显著提升CO2光还原活性

在过去的几十年里,化石燃料燃烧产生的过量二氧化碳(CO2)导致了严重的环境污染,这需要高效的技术将CO2转化为增值化学品和燃料。然而,CO2是一种非极性和高度稳定的分子,需要较高的外部能量来激活和打破强C=O键。此外,大多数CO2加氢反应是通过在高温高压下使用氢气(H2)完成的,而利用地球上丰富的水作为氢源还原CO2在实际应用具有成本效益。
因此,太阳光驱动的光催化被认为是在温和条件下将CO2和水直接转化为含碳化学物质的有效策略。在各种产品中,光催化CO2还原为具有高活性和选择性的CO是一种非常有前景的工艺,实现这一转化过程的关键是开发高效的光催化剂,同时该催化剂应具有良好的电荷分离/转移能力,以及丰富的CO2吸附、活化和质子化活性位点。
尽管人们在光催化剂的设计和制造上做了大量的努力,但在大多数CO2还原光催化剂仍然存在相对较差的活性和选择性。因此,到目前为止,显著提高光催化剂的催化活性和选择性,以实现高效的CO2转化为CO仍然是一个挑战。
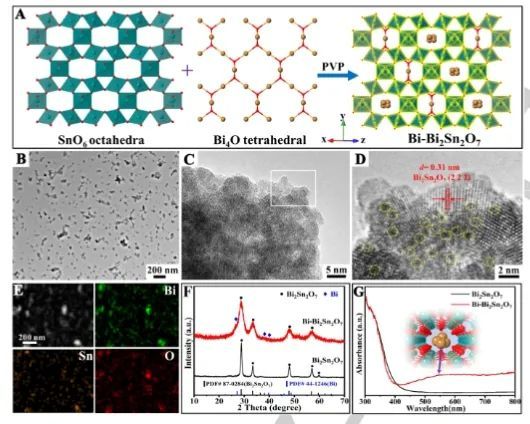
近日,中国科学院兰州化学物理研究所毕迎普课题组通过在Bi2Sn2O7中原位构建空间限制的亚纳米Bi金属团簇(Bi-Bi2Sn2O7),显著增强了光催化CO2还原性能。具体而言,这些空间受限的Bi团簇可以显著促进光生电荷分离、表面电子富集、CO2吸附/活化,有效地提高了光催化CO2还原活性。
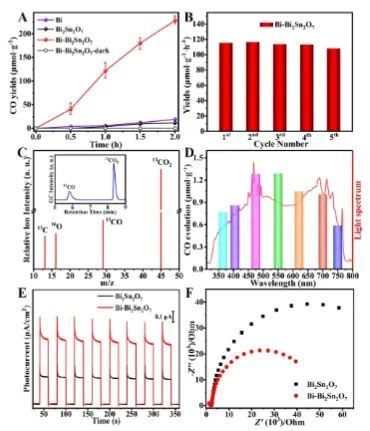
实验结果表明,与纯的Bi2Sn2O7(5.7 μmol g-1 h-1)相比,Bi-Bi2Sn2O7的CO产率显著提高到114.1 μmol g-1 h-1,并且CO的选择性接近100%。同时,Bi-Bi2Sn2O7还表现出优异的稳定性,其在稳定性测试期间活性衰减可忽略不计,并且反应后材料的结构和形貌仍保持良好。

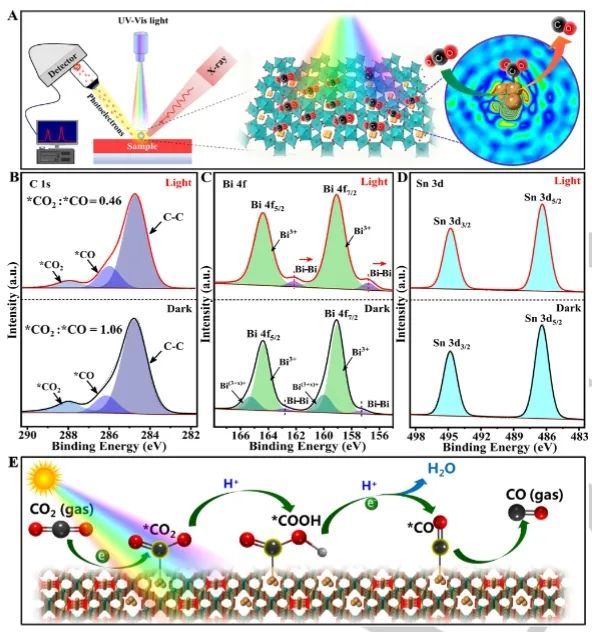
一系列光谱表征和理论计算表明,在光照下,光生电子向Bi团簇流动以产生富电子环境,导致空间受限的金属Bi团簇可以有效促进CO2的吸收和活化,以及加速随后的*CO2质子化形成*COOH中间体;此外,位于Bi团簇上的*COOH可以通过与另一个质子和电子的结合进一步还原,最终实现了*CO解吸形成CO产物。
因此,被限制在Bi2Sn2O7中的Bi金属团簇提供了一个高效的反应通道,以促进光催化CO2还原为CO。总的来说,该项工作不仅突出了限制效应在促进光催化反应方面的巨大潜力,而且为调节金属种类的精确布置和在原子水平上组装特定的限制结构提供了范例。
Subnanometric bismuth clusters confined in pyrochlore-Bi2Sn2O7 enable remarkable CO2 photoreduction. Angewandte Chemie International Edition, 2023. DOI: 10.1002/anie.202316459
7. EES: Ir-O-Ni活性中心起大作用,助力IrOx/Ni(OH)2上UOR/OER灵活转换

电化学制氢是将间歇性可再生能源转化为能够长期储存和长距离运输能源的一种有吸引力的途径。氢气可以通过电解水、生物质资源(酒精、葡萄糖、木质素等)或工业废水(尿素、联氨等)产生。其中,与析氧反应(OER)的高电位(1.27 VRHE)相比,仅需一个小的电位就能驱动尿素氧化反应(UOR)。因此,利用UOR替代水分解反应中的OER可以实现产氢能耗的显著降低。
目前,对于UOR催化剂的研究都是基于尿素浓度恒定的条件下来提高材料的性能。然而,在实际应用中,尿素的浓度并不是不变的,尿素的浓度的波动对制氢的效率有很大的影响,因此,在尿素浓度波动的环境中实现稳定和高效的氢气生产是一个主要的挑战。为了克服这一问题,有必要开发出对UOR和OER都有效的双功能材料,该材料可以随着尿素浓度的变化切换UOR和OER,从而实时适应尿素的浓度波动。


近日,南京大学闫世成课题组Ni(OH)2纳米薄片上电化学沉积均匀的非晶态IrOx纳米薄片,(IrOx/Ni(OH)2),并将其作为高效的OER/UOR双功能电催化剂。电化学性能测试结果显示,IrOx/Ni(OH)2电极在100 mA cm−2电流密度下的OER电位为1.48 VRHE,而在相同电流密度下仅需1.36 VRHE就能驱动UOR;并且,该催化剂在10 mA cm−2电流密度能够连续运行200小时,表现出良好的稳定性。
一系列光谱表征结果显示,在IrOx/Ni(OH)2材料中,Ir-O-Ni活性中心可以适应尿素浓度的波动,改变电子转移方向,从而在UOR和OER之间切换,以实现连续稳定产氢。



具体而言,OER和UOR的活性位点分别是Ir和Ni位点,加入低含量的尿素会吸附在Ir位点上,轻微抑制IrOx/Ni(OH)2的OER电流。在尿素浓度高于0.0825 M(临界尿素浓度)时,OER会转化为UOR。在临界尿素浓度以下,催化剂上尿素分子的覆盖度太低,UOR过程完全受到尿素分子的扩散限制;在临界尿素浓度以上,尿素分子的覆盖度足以打破扩散极限,从而将OER完全转换为UOR。
相应地,电子从Ir位点转移到Ni位点以进行OER或电子从Ni位点转移到Ir位点以进行UOR。电子转移途径的切换是由于IrOx/Ni(OH)2上UOR和OER活性中心的不同,尿素的优先吸附改变了Ni-O-Ir构型的局部电子环境,从而改变了电子转移方向。总之,该项工作所提出的自适应主动中心开关的设计理念为开发在各种工业废水处理系统等复杂介质中连续制氢的催化剂提供了思路。
Reagent-adaptive active site switching on IrOx/Ni(OH)2 catalyst. Energy & Environmental Science, 2023. DOI: 10.1039/D3EE02714G
8. ACS Nano: Fe2P-Co2P异质结构立大功,助力构建高效耐用阴离子交换膜水电解槽

电化学水分解制高纯氢气被认为是缓解能源危机和环境污染问题最有前景的策略。其中,阴离子交换膜水电解槽(AEMWEs)是生产高纯氢气的前沿技术。Pt基材料通常被认为是水电解反应中析氢反应(HER)的基准电催化剂,但其昂贵的价格和稀缺性限制了它们的大规模应用。尽管人们开发了多种非贵金属催化剂,但它们的活性和耐久性(特别是在安培级电流密度的情况下)仍不够理想。因此,开发高活性、高稳定性的非Pt碱性HER催化剂以满足工业AEM在安培电流下具有低过电位和长期使用寿命的需要,是推进AEM电解槽实际应用的关键。


近日,云南大学胡广志课题组通过配体交换调控策略合成了锚定在N和P双掺杂碳多孔纳米片上的超细Fe2P和Co2P纳米颗粒(Fe2P-Co2P/NPC)。理论计算表明,与Fe2P/NPC和Co2P/NPC中电子分别在Fe和Co位点富集不同,Fe2P-Co2P/NPC中的电子从Fe和Co原子流向P原子,使得电子在P位点重新分配和富集。
这表明Fe2P-Co2P/NPC在快速配体交换反应后有效地优化了Fe和Ni原子的电子状态,显著提高了其催化活性。同时,电子从Fe和Co向P的逆向流动激活了Fe2P-Co2P/NPC异质结中界面Co-P-Fe桥联活性位点,优化了氢中间体的吸附和脱附,从而产生更有利的HER动力学。
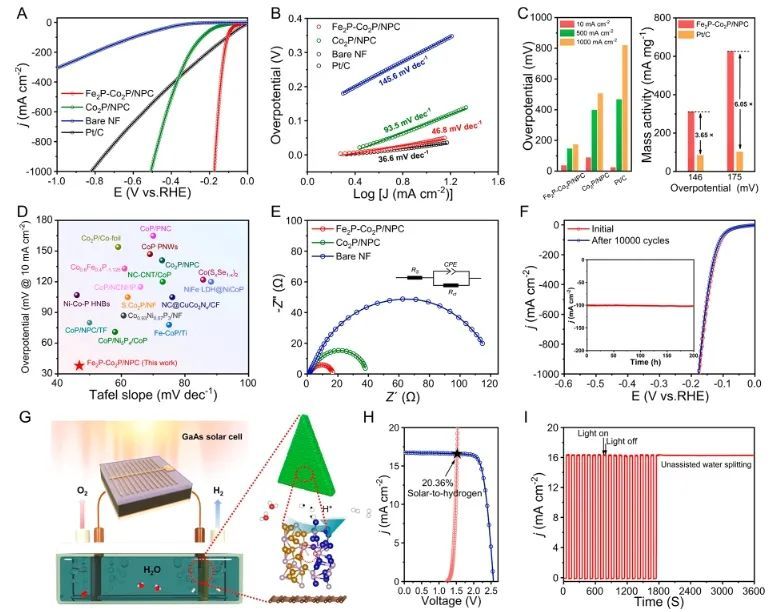

因此,所制备的Fe2P-Co2P/NPC催化剂在碱性条件下分别仅需38、147和175 mV的过电位就能达到10、500和1000 mA cm−2的电流密度;同时,该催化剂在100 mA cm−2电流密度下连续运行200小时而没有发生明显的活性下降,且反应后材料的结构和形貌保持良好,表明其具有优异的稳定性。
更重要的是,利用Fe2P-Co2P/NPC作为阴极组装的阴离子交换膜(AEM)水电解槽(Fe2P-Co2P/NPC||NiFe (OH)x-Ni3S2),仅需1.73 V的电池电压就能产生1000 mA cm−2的电流密度,并且该电解槽在1000 mA cm−2电流密度下连续运行1000小时而没有发生活性衰减,表现出巨大的实际应用潜力。
简而言之,该项工作为开发高活性、高稳定性的非贵金属催化剂用于工业规模的清洁能源生产提供了一种高效可行的策略。
MOF-on-MOF-derived ultrafine Fe2P-Co2P heterostructures for high-efficiency and durable anion exchange membrane water electrolyzers. ACS Nano, 2023. DOI: 10.1021/acsnano.3c09020
