
电催化水分解被认为是一种可持续的、生态友好的生产清洁氢能的有效途径。然而,水电解的阳极析氧反应(OER)是一个复杂的四电子转移过程,动力学过程缓慢,限制了水电解制氢的整体效率。RuO2和IrO2等贵金属基催化剂表现出优异的OER催化性能,但其稀缺性和高成本限制了它们在水电解中的大规模应用。因此,开发高效且具有成本效益的非贵金属基电催化剂以推动水电解制氢技术的实际应用具有重要意义。
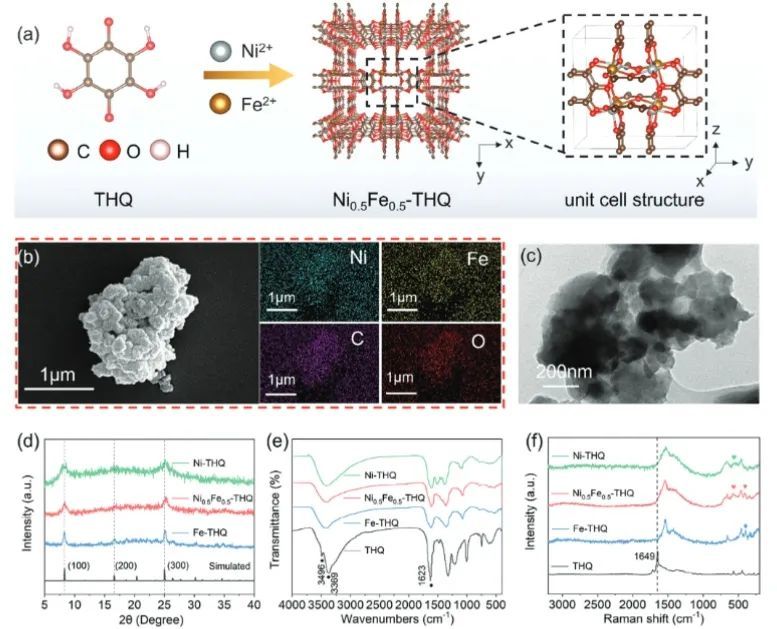

近日,武汉科技大学杜星和中国科学院化学研究所董际臣等报道了一种利用具有高密度催化活性位点的最小苯基配体制备一系列双金属c-MOFs (NixFe1-x-THQ,THQ=四羟基-1,4-苯醌水合物)的简便方法。通过调节Ni/Fe的比例,得到了一系列具有最佳电子结构和中间体吸附能力、电荷传输速率快的催化剂。
实验结果表明,最优的Ni0.5Fe0.5-THQ催化剂在10 mA cm−2电流密度下的OER过电位仅为272 mV,并且该催化剂在1.5 VRHE下连续运行40小时而没有发生明显的活性下降,且反应后材料的形貌和结构没有发生变化,表明Ni0.5Fe0.5-THQ具有优异的稳定性(这种显著的电化学稳定性可以归因于Ni、Fe和THQ之间配位键的强螯合作用)。


一系列光谱表征和理论计算结果显示,由于具有高自旋价电子排布和π对称(t2g) d轨道中相应的三个未配对电子,Fe3+ (t2g3eg2)可以实现与桥接O之间较弱的电子相互作用,而Ni2+ (t2g6eg2)可以实现与桥接O之间强烈的e−-e−排斥,因为其具有完全占据π对称(t2g)d-轨道。
因此,当Ni2+与Fe3+耦合后,O2-与Ni2+之间强烈的e−-e−排斥作用将触发Ni2+向Fe3+的部分电子转移,进而很好地调制NixFe1-x-THQ中Ni位点的电子构型,使得Ni位点与吸附氧物种之间的结合强度可以优化到满足Sabatier原则。
此外,随着电子密度的降低,具有低半满eg轨道的Ni2+与吸附的氧物种形成适当的键合,有利于提高催化活性,加快OER反应速率。综上,该项工作为构建具有更多高密度催化活性中心的双金属c-MOFs电催化剂以实现高效水电解提供了指导,也为MOFs电催化剂的设计和研究提供了新的思路。
Regulating electronic structure of bimetallic NiFe-THQ conductive metal–organic frameworks to boost catalytic activity for oxygen evolution reaction. Advanced Functional Materials, 2023. DOI: 10.1002/adfm.202310902
