
合成致死性最初是在果蝇中观察到的,定义为单个可存活基因共同改变的结果。合成致死已成为一种很有前景的抗癌药物发现策略,尤其是针对一些有挑战性的肿瘤抑制基因。通过利用合成致死的机制,可以有选择地靶向具有突变或失活的驱动基因的癌细胞,潜在地减少治疗的副作用[1]。在同源重组修复缺陷型的肿瘤患者中使用聚腺苷二磷酸核糖聚合酶(PARP)抑制剂是靶向治疗的经典例子,亦是“合成致死”效应成功转化为临床应用的典型代表之一。迄今为止,这种治疗方法已成功在卵巢癌、乳腺癌、前列腺癌和胰腺癌中获批,其潜在的治疗获益也正在子宫内膜癌、肾癌、胃肠道等肿瘤中被挖掘和证实[2]。然而,一项最新的研究结论可能会颠覆此前已知的PARP抑制剂作用机制,该研究结果认为在同源重组修复缺陷的癌细胞中,PARP抑制剂杀死癌细胞的方式是由于转录-复制冲突(TRCs)而非“捕获”。
PARP抑制剂已知的作用机制
PARP是一种核酶,参与多种DNA修复途径,其中研究最广泛的是对于核苷酸切除修复(BER)的作用。目前已鉴定出17个PARP家族成员,但只有PARP1,PARP2在DNA修复中发挥作用。PARP在翻译后改变自身和/或具有负电荷的多(ADP-核糖)(PAR)部分的其他蛋白质,这个过程被称为PARylation(PAR修饰)。PARP1承担了此过程中80%的工作。PARP抑制剂的作用机制,传统多认可的是“捕获”学说,即:①在PARP活性位点与烟酰胺腺嘌呤二核苷酸竞争,抑制多聚(ADP-核糖)聚合物形成;②结合到PARP1和/或PARP2的烟酰胺腺嘌呤二核苷酸结合口袋,造成构象异构,稳定DNA-PARP的可逆解离,使PARP保持对DNA的结合,这一过程被称为DNA-PARP复合物的“捕获”,从而导致DNA-PARP复合物长期存在,抑制DNA后续修复过程[3]。

PARP抑制剂及其可能的作用机制[4]
HRD肿瘤的“合成致死”效应
当细胞遭受DNA损伤时,出现单链断裂。PARPs,主要是PARP1与DNA断裂位点结合,发生构象变化,从而增加其催化活性。由此产生的PADPr聚合物改变了被修饰蛋白质的功能(例如降低了PARP1对受损DNA的亲和力,使其解离),并招募其他蛋白质非共价的结合到聚合物上。用PARP抑制剂治疗HRD的肿瘤患者时,PARP抑制剂将PARP捕捉在DNA上。被捕获的PARP会阻止复制叉的进展,从而导致DNA双链断裂的形成。此时,NHEJ是修复双链断裂的唯一途径,这会导致基因组不稳定和细胞死亡[5]。
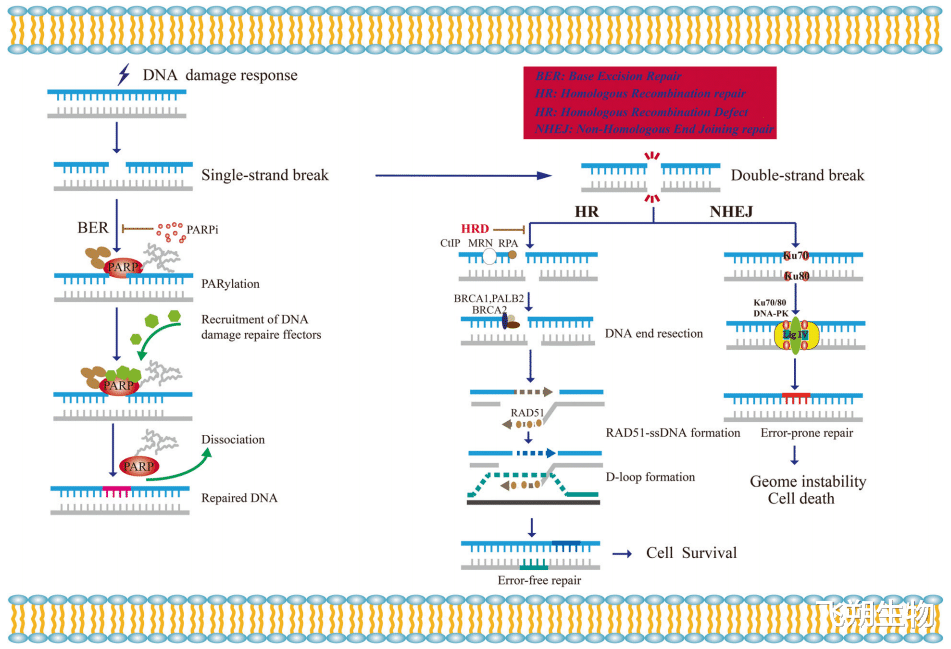
PARP和HRD之间的合成致死相互作用[6]
PARP抑制剂作用机制的新的发现
基于上述已知原理,原则上PARP抑制剂的抑制能力应与PARPs被捕获的能力成正比。但是有些研究发现PARP抑制剂对PARPs的抑制能力与其对PARPs的捕获能力并无很好的相关性。这似乎说明其“合成致死”效应并非表面上看起来这么简单。
细胞内转录和复制都需要大分子复合物作用于DNA模板,然而这些功能不能同时作用于同一DNA序列。复制和转录机制之间的冲突(转录-复制冲突,TRCs)在原核生物和真核生物中都广泛存在,并且能够导致DNA损伤、基因组的不稳定、影响高保真的复制[7]。
TIMELESS和TIPIN是两个可保护复制体不与转录冲突的蛋白质,体外实验表明PARP抑制剂的治疗,特别是在S期早期,可导致TRCs。
为了进一步探索诱导TRCs依赖性DNA损伤反应是否需要PARP捕获,研究者通过siRNA去除了PARP1和PARP2,并监测了S期HeLa细胞中的DNA损伤标记物。PARP1的缺失以依赖于转录延伸的方式诱导了DNA损伤反应,而PARP2的缺失则没有影响。该结果支持以下结论,即:抑制PARP酶活性足以诱导TRCs,导致DNA损伤,而与捕获无关,因为去除的PARP不能被捕获。同时表明似乎只有PARP1具有防止TRCs诱导的DNA损伤的功能。
已知PARP1和TIMELESS相互影响,这表明它们可能通过相同的分子途径来阻止TRCs。研究者在PARP抑制剂处理的细胞中用siRNA敲除TIMELESS或TIPIN。TIMELESS或TIPIN的缺失导致TRCs无法避免,PARP1的活性增强。TIMELESS和PARP1蛋白的表达阻止了TRCs和TRCs诱导的DNA损伤。
同时发现PARP1的缺失加速了复制叉的发展,其程度与TIMELESS或TIPIN的缺失相似,而PARP2的缺失没有明显的影响,此结果与PARP1通过TIMELESS向复制体传递TRCs信号的结论一致,而PARP2不与TIMELESS相互作用。
因为PARP1、TIMELESS和TIPIN在预防TRCs的相同途径中发挥作用,研究者检测了TIMELESS或TIPIN的缺失是否会与HRD细胞形成“合成致死”。BRCA2和TIMELESS或BRCA2和TIPIN的共缺失引起HeLa细胞中强烈的DNA损伤反应,并导致细胞死亡。这可能对药物的研发提供新的靶点。
总的来说,PARP1向TIMELESS和TIPIN发出即将出现TRCs的信号,于是细胞暂停复制直到TRCs被解离。如果PARP1或TIMELESS和TIPIN未能发挥其功能,TRCs就会导致DNA损伤,需要HR进行修复。PARP抑制剂新旧机制之间的主要区别是复制体冲突对象的性质:被捕获的PARPs或转录延伸复合物。支持新机制的依据是:HRD细胞中与PARP抑制剂的合成致死性可以通过抑制转录延伸来减轻,并且siRNA耗尽TIMELESS、TIPIN或PARP1对HRD是合成致死性的[8]。
新机制的发现有助于临床药物的开发
加深对PARP抑制剂如何靶向HRD细胞的理解,有助于指导其未来的临床开发。目前在临床上使用的所有PARP抑制剂都抑制PARP1和PARP2。然而,可能只有PARP1保护复制体不受TRCs的影响。因此,PARP1选择性抑制剂,如saruparib,可能足以在临床上诱导HRD的合成致死性。新机制同时提出了一个问题,即调节PARP捕获能力是否可以增强PARP抑制剂在临床上的应用可及性。PARP抑制剂的捕获能力不同,因此这是一个可以独立于抑制能力进行优化的参数。研究结论认为降低捕获力可能会在不影响疗效的前提下降低PARP抑制剂的毒性。
参考文献:
J Med Chem 2024 Jul 25;67(14):11488-11521.
Nat Commun 2020 Nov 4;11(1):5584.
卵巢癌PARP抑制剂临床应用指南
Nat Rev Clin Oncol 2021 Dec;18(12):773-791.
J Clin Oncol 2015 Apr 20;33(12):1397-406.
Mol Cancer 2020 Jun 20;19(1):107.
Annu Rev Genet2023 Nov 27:57:157-179.
Nature 2024 Apr;628(8007):433-441.
